Federico Catalano, Thomas J O'Brien, Aleksandra A Mekhova, Lucia Vittoria Sepe, Mariantonietta Elia, Rossella De Cegli, Ivan Gallotta, Pamela Santonicola, Giuseppina Zampi, Ekaterina Y Ilyechova, Aleksei A Romanov, Polina D Samuseva, Josephine Salzano, Raffaella Petruzzelli, Elena V Polishchuk, Alessia Indrieri, Byung-Eun Kim, André E X Brown, Ludmila V Puchkova, Elia Di Schiavi, Roman S Polishchuk
{"title":"一种新的秀丽隐杆线虫模型,用于研究Wilson病中铜的毒性。","authors":"Federico Catalano, Thomas J O'Brien, Aleksandra A Mekhova, Lucia Vittoria Sepe, Mariantonietta Elia, Rossella De Cegli, Ivan Gallotta, Pamela Santonicola, Giuseppina Zampi, Ekaterina Y Ilyechova, Aleksei A Romanov, Polina D Samuseva, Josephine Salzano, Raffaella Petruzzelli, Elena V Polishchuk, Alessia Indrieri, Byung-Eun Kim, André E X Brown, Ludmila V Puchkova, Elia Di Schiavi, Roman S Polishchuk","doi":"10.1111/tra.12920","DOIUrl":null,"url":null,"abstract":"<p><p>Wilson disease (WD) is caused by mutations in the ATP7B gene that encodes a copper (Cu) transporting ATPase whose trafficking from the Golgi to endo-lysosomal compartments drives sequestration of excess Cu and its further excretion from hepatocytes into the bile. Loss of ATP7B function leads to toxic Cu overload in the liver and subsequently in the brain, causing fatal hepatic and neurological abnormalities. The limitations of existing WD therapies call for the development of new therapeutic approaches, which require an amenable animal model system for screening and validation of drugs and molecular targets. To achieve this objective, we generated a mutant Caenorhabditis elegans strain with a substitution of a conserved histidine (H828Q) in the ATP7B ortholog cua-1 corresponding to the most common ATP7B variant (H1069Q) that causes WD. cua-1 mutant animals exhibited very poor resistance to Cu compared to the wild-type strain. This manifested in a strong delay in larval development, a shorter lifespan, impaired motility, oxidative stress pathway activation, and mitochondrial damage. In addition, morphological analysis revealed several neuronal abnormalities in cua-1 mutant animals exposed to Cu. Further investigation suggested that mutant CUA-1 is retained and degraded in the endoplasmic reticulum, similarly to human ATP7B-H1069Q. As a consequence, the mutant protein does not allow animals to counteract Cu toxicity. Notably, pharmacological correctors of ATP7B-H1069Q reduced Cu toxicity in cua-1 mutants indicating that similar pathogenic molecular pathways might be activated by the H/Q substitution and, therefore, targeted for rescue of ATP7B/CUA-1 function. Taken together, our findings suggest that the newly generated cua-1 mutant strain represents an excellent model for Cu toxicity studies in WD.</p>","PeriodicalId":23207,"journal":{"name":"Traffic","volume":" ","pages":"e12920"},"PeriodicalIF":2.8000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10841361/pdf/","citationCount":"0","resultStr":"{\"title\":\"A new Caenorhabditis elegans model to study copper toxicity in Wilson disease.\",\"authors\":\"Federico Catalano, Thomas J O'Brien, Aleksandra A Mekhova, Lucia Vittoria Sepe, Mariantonietta Elia, Rossella De Cegli, Ivan Gallotta, Pamela Santonicola, Giuseppina Zampi, Ekaterina Y Ilyechova, Aleksei A Romanov, Polina D Samuseva, Josephine Salzano, Raffaella Petruzzelli, Elena V Polishchuk, Alessia Indrieri, Byung-Eun Kim, André E X Brown, Ludmila V Puchkova, Elia Di Schiavi, Roman S Polishchuk\",\"doi\":\"10.1111/tra.12920\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Wilson disease (WD) is caused by mutations in the ATP7B gene that encodes a copper (Cu) transporting ATPase whose trafficking from the Golgi to endo-lysosomal compartments drives sequestration of excess Cu and its further excretion from hepatocytes into the bile. Loss of ATP7B function leads to toxic Cu overload in the liver and subsequently in the brain, causing fatal hepatic and neurological abnormalities. The limitations of existing WD therapies call for the development of new therapeutic approaches, which require an amenable animal model system for screening and validation of drugs and molecular targets. To achieve this objective, we generated a mutant Caenorhabditis elegans strain with a substitution of a conserved histidine (H828Q) in the ATP7B ortholog cua-1 corresponding to the most common ATP7B variant (H1069Q) that causes WD. cua-1 mutant animals exhibited very poor resistance to Cu compared to the wild-type strain. This manifested in a strong delay in larval development, a shorter lifespan, impaired motility, oxidative stress pathway activation, and mitochondrial damage. In addition, morphological analysis revealed several neuronal abnormalities in cua-1 mutant animals exposed to Cu. Further investigation suggested that mutant CUA-1 is retained and degraded in the endoplasmic reticulum, similarly to human ATP7B-H1069Q. As a consequence, the mutant protein does not allow animals to counteract Cu toxicity. Notably, pharmacological correctors of ATP7B-H1069Q reduced Cu toxicity in cua-1 mutants indicating that similar pathogenic molecular pathways might be activated by the H/Q substitution and, therefore, targeted for rescue of ATP7B/CUA-1 function. Taken together, our findings suggest that the newly generated cua-1 mutant strain represents an excellent model for Cu toxicity studies in WD.</p>\",\"PeriodicalId\":23207,\"journal\":{\"name\":\"Traffic\",\"volume\":\" \",\"pages\":\"e12920\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2024-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10841361/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Traffic\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1111/tra.12920\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/10/27 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Traffic","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1111/tra.12920","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/10/27 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
A new Caenorhabditis elegans model to study copper toxicity in Wilson disease.
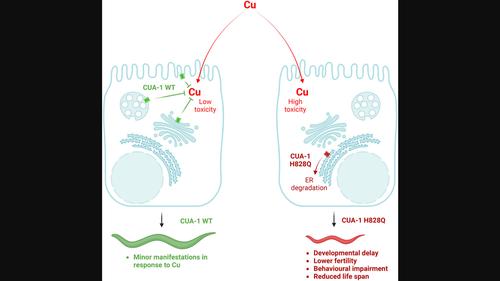
Wilson disease (WD) is caused by mutations in the ATP7B gene that encodes a copper (Cu) transporting ATPase whose trafficking from the Golgi to endo-lysosomal compartments drives sequestration of excess Cu and its further excretion from hepatocytes into the bile. Loss of ATP7B function leads to toxic Cu overload in the liver and subsequently in the brain, causing fatal hepatic and neurological abnormalities. The limitations of existing WD therapies call for the development of new therapeutic approaches, which require an amenable animal model system for screening and validation of drugs and molecular targets. To achieve this objective, we generated a mutant Caenorhabditis elegans strain with a substitution of a conserved histidine (H828Q) in the ATP7B ortholog cua-1 corresponding to the most common ATP7B variant (H1069Q) that causes WD. cua-1 mutant animals exhibited very poor resistance to Cu compared to the wild-type strain. This manifested in a strong delay in larval development, a shorter lifespan, impaired motility, oxidative stress pathway activation, and mitochondrial damage. In addition, morphological analysis revealed several neuronal abnormalities in cua-1 mutant animals exposed to Cu. Further investigation suggested that mutant CUA-1 is retained and degraded in the endoplasmic reticulum, similarly to human ATP7B-H1069Q. As a consequence, the mutant protein does not allow animals to counteract Cu toxicity. Notably, pharmacological correctors of ATP7B-H1069Q reduced Cu toxicity in cua-1 mutants indicating that similar pathogenic molecular pathways might be activated by the H/Q substitution and, therefore, targeted for rescue of ATP7B/CUA-1 function. Taken together, our findings suggest that the newly generated cua-1 mutant strain represents an excellent model for Cu toxicity studies in WD.
期刊介绍:
Traffic encourages and facilitates the publication of papers in any field relating to intracellular transport in health and disease. Traffic papers span disciplines such as developmental biology, neuroscience, innate and adaptive immunity, epithelial cell biology, intracellular pathogens and host-pathogen interactions, among others using any eukaryotic model system. Areas of particular interest include protein, nucleic acid and lipid traffic, molecular motors, intracellular pathogens, intracellular proteolysis, nuclear import and export, cytokinesis and the cell cycle, the interface between signaling and trafficking or localization, protein translocation, the cell biology of adaptive an innate immunity, organelle biogenesis, metabolism, cell polarity and organization, and organelle movement.
All aspects of the structural, molecular biology, biochemistry, genetics, morphology, intracellular signaling and relationship to hereditary or infectious diseases will be covered. Manuscripts must provide a clear conceptual or mechanistic advance. The editors will reject papers that require major changes, including addition of significant experimental data or other significant revision.
Traffic will consider manuscripts of any length, but encourages authors to limit their papers to 16 typeset pages or less.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
