{"title":"有条件从头药物设计的图神经网络","authors":"Carlo Abate, Sergio Decherchi, Andrea Cavalli","doi":"10.1002/wcms.1651","DOIUrl":null,"url":null,"abstract":"<p>Drug design is costly in terms of resources and time. Generative deep learning techniques are using increasing amounts of biochemical data and computing power to pave the way for a new generation of tools and methods for drug discovery and optimization. Although early methods used SMILES strings, more recent approaches use molecular graphs to naturally represent chemical entities. Graph neural networks (GNNs) are learning models that can natively process graphs. The use of GNNs in drug discovery is growing exponentially. GNNs for drug design are often coupled with conditioning techniques to steer the generation process towards desired chemical and biological properties. These conditioned graph-based generative models and frameworks hold promise for the routine application of GNNs in drug discovery.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"13 4","pages":""},"PeriodicalIF":27.0000,"publicationDate":"2023-01-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":"{\"title\":\"Graph neural networks for conditional de novo drug design\",\"authors\":\"Carlo Abate, Sergio Decherchi, Andrea Cavalli\",\"doi\":\"10.1002/wcms.1651\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Drug design is costly in terms of resources and time. Generative deep learning techniques are using increasing amounts of biochemical data and computing power to pave the way for a new generation of tools and methods for drug discovery and optimization. Although early methods used SMILES strings, more recent approaches use molecular graphs to naturally represent chemical entities. Graph neural networks (GNNs) are learning models that can natively process graphs. The use of GNNs in drug discovery is growing exponentially. GNNs for drug design are often coupled with conditioning techniques to steer the generation process towards desired chemical and biological properties. These conditioned graph-based generative models and frameworks hold promise for the routine application of GNNs in drug discovery.</p><p>This article is categorized under:\\n </p>\",\"PeriodicalId\":236,\"journal\":{\"name\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"volume\":\"13 4\",\"pages\":\"\"},\"PeriodicalIF\":27.0000,\"publicationDate\":\"2023-01-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1651\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1651","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Graph neural networks for conditional de novo drug design
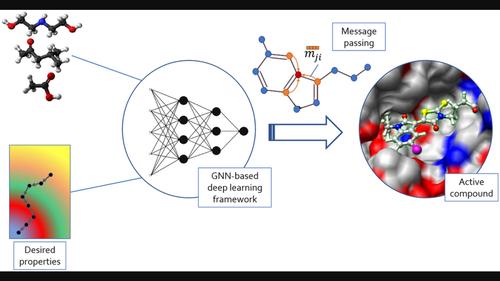
Drug design is costly in terms of resources and time. Generative deep learning techniques are using increasing amounts of biochemical data and computing power to pave the way for a new generation of tools and methods for drug discovery and optimization. Although early methods used SMILES strings, more recent approaches use molecular graphs to naturally represent chemical entities. Graph neural networks (GNNs) are learning models that can natively process graphs. The use of GNNs in drug discovery is growing exponentially. GNNs for drug design are often coupled with conditioning techniques to steer the generation process towards desired chemical and biological properties. These conditioned graph-based generative models and frameworks hold promise for the routine application of GNNs in drug discovery.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
