Michele Nottoli, Mattia Bondanza, Patrizia Mazzeo, Lorenzo Cupellini, Carles Curutchet, Daniele Loco, Louis Lagardère, Jean-Philip Piquemal, Benedetta Mennucci, Filippo Lipparini
{"title":"QM/AMOEBA对嵌入分子性质和动力学的描述","authors":"Michele Nottoli, Mattia Bondanza, Patrizia Mazzeo, Lorenzo Cupellini, Carles Curutchet, Daniele Loco, Louis Lagardère, Jean-Philip Piquemal, Benedetta Mennucci, Filippo Lipparini","doi":"10.1002/wcms.1674","DOIUrl":null,"url":null,"abstract":"<p>We describe the development, implementation, and application of a polarizable QM/MM strategy, based on the AMOEBA polarizable force field, for calculating molecular properties and performing dynamics of molecular systems embedded in complex matrices. We show that polarizable QM/MM is a well-understood, mature technology that can be deployed using a state-of-the-art implementation that combines efficient numerical methods and linear scaling techniques. Thanks to these numerical advances and to the availability of parameters for a wide manifold of systems in the AMOEBA force field, polarizable QM/AMOEBA can be used for advanced production applications, that range from the prediction of spectroscopies to ground- and excited-state multiscale ab initio molecular dynamics simulations.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"13 6","pages":""},"PeriodicalIF":27.0000,"publicationDate":"2023-06-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"2","resultStr":"{\"title\":\"QM/AMOEBA description of properties and dynamics of embedded molecules\",\"authors\":\"Michele Nottoli, Mattia Bondanza, Patrizia Mazzeo, Lorenzo Cupellini, Carles Curutchet, Daniele Loco, Louis Lagardère, Jean-Philip Piquemal, Benedetta Mennucci, Filippo Lipparini\",\"doi\":\"10.1002/wcms.1674\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>We describe the development, implementation, and application of a polarizable QM/MM strategy, based on the AMOEBA polarizable force field, for calculating molecular properties and performing dynamics of molecular systems embedded in complex matrices. We show that polarizable QM/MM is a well-understood, mature technology that can be deployed using a state-of-the-art implementation that combines efficient numerical methods and linear scaling techniques. Thanks to these numerical advances and to the availability of parameters for a wide manifold of systems in the AMOEBA force field, polarizable QM/AMOEBA can be used for advanced production applications, that range from the prediction of spectroscopies to ground- and excited-state multiscale ab initio molecular dynamics simulations.</p><p>This article is categorized under:\\n </p>\",\"PeriodicalId\":236,\"journal\":{\"name\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"volume\":\"13 6\",\"pages\":\"\"},\"PeriodicalIF\":27.0000,\"publicationDate\":\"2023-06-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1674\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1674","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
QM/AMOEBA description of properties and dynamics of embedded molecules
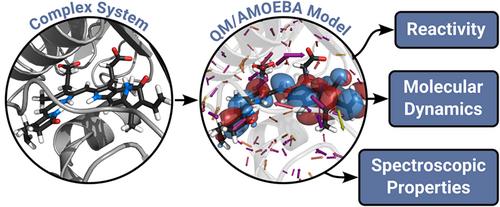
We describe the development, implementation, and application of a polarizable QM/MM strategy, based on the AMOEBA polarizable force field, for calculating molecular properties and performing dynamics of molecular systems embedded in complex matrices. We show that polarizable QM/MM is a well-understood, mature technology that can be deployed using a state-of-the-art implementation that combines efficient numerical methods and linear scaling techniques. Thanks to these numerical advances and to the availability of parameters for a wide manifold of systems in the AMOEBA force field, polarizable QM/AMOEBA can be used for advanced production applications, that range from the prediction of spectroscopies to ground- and excited-state multiscale ab initio molecular dynamics simulations.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
