Wan-Ping Lee , Qihui Zhu , Xiaofei Yang , Silvia Liu , Eliza Cerveira , Mallory Ryan , Adam Mil-Homens , Lauren Bellfy , Kai Ye , Charles Lee , Chengsheng Zhang
{"title":"JAX-CNV:一种基于全基因组测序的临床级拷贝数检测算法","authors":"Wan-Ping Lee , Qihui Zhu , Xiaofei Yang , Silvia Liu , Eliza Cerveira , Mallory Ryan , Adam Mil-Homens , Lauren Bellfy , Kai Ye , Charles Lee , Chengsheng Zhang","doi":"10.1016/j.gpb.2021.06.003","DOIUrl":null,"url":null,"abstract":"<div><p>We aimed to develop a <strong>whole-genome sequencing</strong> (WGS)-based <strong>copy number variant</strong> (CNV) calling algorithm with the potential of replacing <strong>chromosomal microarray assay</strong> (CMA) for clinical diagnosis. <strong>JAX-CNV</strong> is thus developed for CNV detection from WGS data. The performance of this CNV calling algorithm was evaluated in a blinded manner on 31 samples and compared to the 112 CNVs reported by clinically validated CMAs for these 31 samples. The result showed that JAX-CNV recalled 100% of these CNVs. Besides, JAX-CNV identified an average of 30 CNVs per individual, respresenting an approximately seven-fold increase compared to calls of clinically validated CMAs. Experimental validation of 24 randomly selected CNVs showed one false positive, <em>i.e.</em>, a false discovery rate (FDR) of 4.17%. A robustness test on lower-coverage data revealed a 100% sensitivity for CNVs larger than 300 kb (the current threshold for College of American Pathologists) down to 10× coverage. For CNVs larger than 50 kb, sensitivities were 100% for coverages deeper than 20×, 97% for 15×, and 95% for 10×. We developed a WGS-based CNV pipeline, including this newly developed CNV caller JAX-CNV, and found it capable of detecting CMA-reported CNVs at a sensitivity of 100% with about a FDR of 4%. We propose that JAX-CNV could be further examined in a multi-institutional study to justify the transition of first-tier <strong>genetic testing</strong> from CMAs to WGS. JAX-CNV is available at <span>https://github.com/TheJacksonLaboratory/JAX-CNV</span><svg><path></path></svg>.</p></div>","PeriodicalId":12528,"journal":{"name":"Genomics, Proteomics & Bioinformatics","volume":"20 6","pages":"Pages 1197-1206"},"PeriodicalIF":7.9000,"publicationDate":"2022-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10225484/pdf/","citationCount":"0","resultStr":"{\"title\":\"JAX-CNV: A Whole-genome Sequencing-based Algorithm for Copy Number Detection at Clinical Grade Level\",\"authors\":\"Wan-Ping Lee , Qihui Zhu , Xiaofei Yang , Silvia Liu , Eliza Cerveira , Mallory Ryan , Adam Mil-Homens , Lauren Bellfy , Kai Ye , Charles Lee , Chengsheng Zhang\",\"doi\":\"10.1016/j.gpb.2021.06.003\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>We aimed to develop a <strong>whole-genome sequencing</strong> (WGS)-based <strong>copy number variant</strong> (CNV) calling algorithm with the potential of replacing <strong>chromosomal microarray assay</strong> (CMA) for clinical diagnosis. <strong>JAX-CNV</strong> is thus developed for CNV detection from WGS data. The performance of this CNV calling algorithm was evaluated in a blinded manner on 31 samples and compared to the 112 CNVs reported by clinically validated CMAs for these 31 samples. The result showed that JAX-CNV recalled 100% of these CNVs. Besides, JAX-CNV identified an average of 30 CNVs per individual, respresenting an approximately seven-fold increase compared to calls of clinically validated CMAs. Experimental validation of 24 randomly selected CNVs showed one false positive, <em>i.e.</em>, a false discovery rate (FDR) of 4.17%. A robustness test on lower-coverage data revealed a 100% sensitivity for CNVs larger than 300 kb (the current threshold for College of American Pathologists) down to 10× coverage. For CNVs larger than 50 kb, sensitivities were 100% for coverages deeper than 20×, 97% for 15×, and 95% for 10×. We developed a WGS-based CNV pipeline, including this newly developed CNV caller JAX-CNV, and found it capable of detecting CMA-reported CNVs at a sensitivity of 100% with about a FDR of 4%. We propose that JAX-CNV could be further examined in a multi-institutional study to justify the transition of first-tier <strong>genetic testing</strong> from CMAs to WGS. JAX-CNV is available at <span>https://github.com/TheJacksonLaboratory/JAX-CNV</span><svg><path></path></svg>.</p></div>\",\"PeriodicalId\":12528,\"journal\":{\"name\":\"Genomics, Proteomics & Bioinformatics\",\"volume\":\"20 6\",\"pages\":\"Pages 1197-1206\"},\"PeriodicalIF\":7.9000,\"publicationDate\":\"2022-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10225484/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genomics, Proteomics & Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1672022922000055\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/1/25 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genomics, Proteomics & Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1672022922000055","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/25 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
JAX-CNV: A Whole-genome Sequencing-based Algorithm for Copy Number Detection at Clinical Grade Level
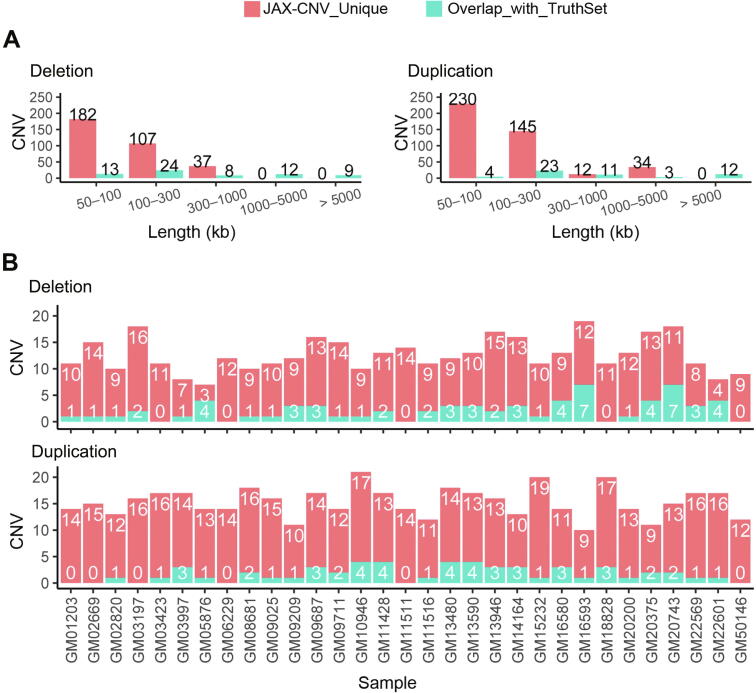
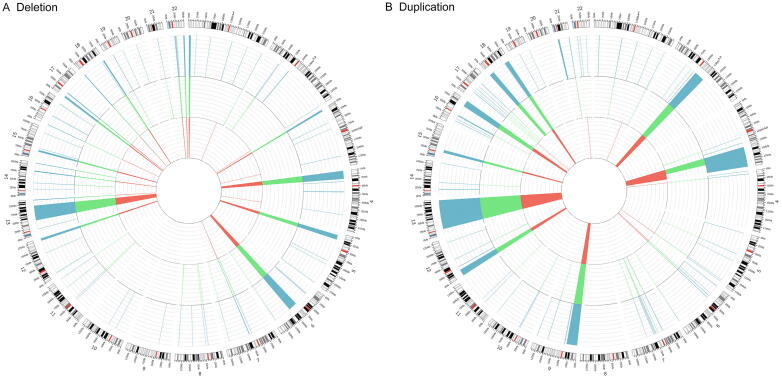
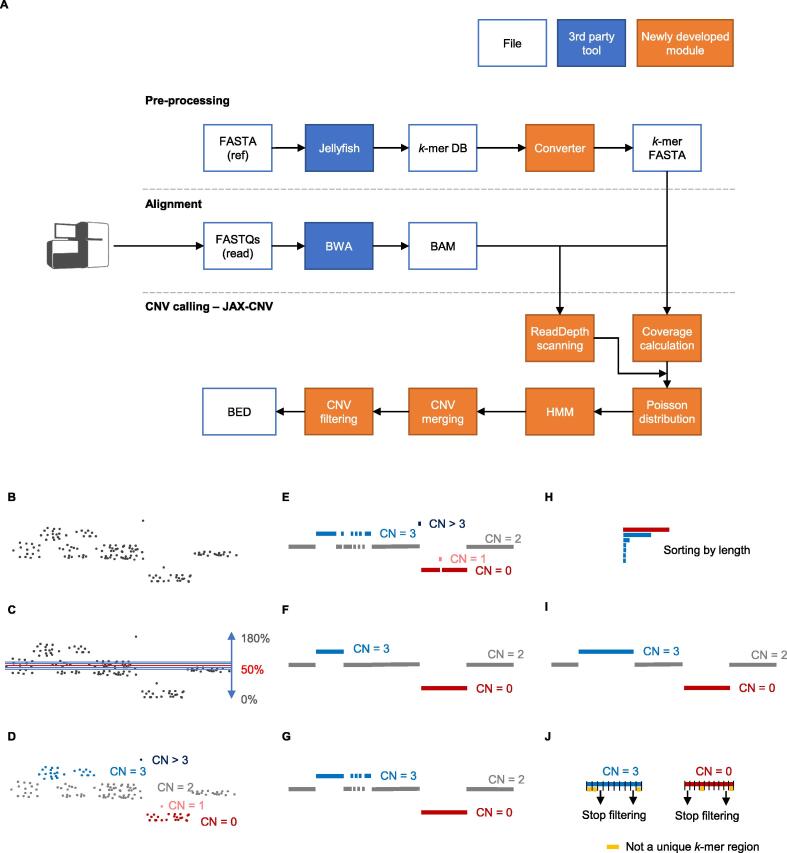
We aimed to develop a whole-genome sequencing (WGS)-based copy number variant (CNV) calling algorithm with the potential of replacing chromosomal microarray assay (CMA) for clinical diagnosis. JAX-CNV is thus developed for CNV detection from WGS data. The performance of this CNV calling algorithm was evaluated in a blinded manner on 31 samples and compared to the 112 CNVs reported by clinically validated CMAs for these 31 samples. The result showed that JAX-CNV recalled 100% of these CNVs. Besides, JAX-CNV identified an average of 30 CNVs per individual, respresenting an approximately seven-fold increase compared to calls of clinically validated CMAs. Experimental validation of 24 randomly selected CNVs showed one false positive, i.e., a false discovery rate (FDR) of 4.17%. A robustness test on lower-coverage data revealed a 100% sensitivity for CNVs larger than 300 kb (the current threshold for College of American Pathologists) down to 10× coverage. For CNVs larger than 50 kb, sensitivities were 100% for coverages deeper than 20×, 97% for 15×, and 95% for 10×. We developed a WGS-based CNV pipeline, including this newly developed CNV caller JAX-CNV, and found it capable of detecting CMA-reported CNVs at a sensitivity of 100% with about a FDR of 4%. We propose that JAX-CNV could be further examined in a multi-institutional study to justify the transition of first-tier genetic testing from CMAs to WGS. JAX-CNV is available at https://github.com/TheJacksonLaboratory/JAX-CNV.
期刊介绍:
Genomics, Proteomics and Bioinformatics (GPB) is the official journal of the Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation and Genetics Society of China. It aims to disseminate new developments in the field of omics and bioinformatics, publish high-quality discoveries quickly, and promote open access and online publication. GPB welcomes submissions in all areas of life science, biology, and biomedicine, with a focus on large data acquisition, analysis, and curation. Manuscripts covering omics and related bioinformatics topics are particularly encouraged. GPB is indexed/abstracted by PubMed/MEDLINE, PubMed Central, Scopus, BIOSIS Previews, Chemical Abstracts, CSCD, among others.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
