Yali Hou , Shilei Zhao , Qi Liu , Xiaolong Zhang , Tong Sha , Yankai Su , Wenming Zhao , Yiming Bao , Yongbiao Xue , Hua Chen
{"title":"持续的正选择驱动SARS-CoV-2基因组的进化","authors":"Yali Hou , Shilei Zhao , Qi Liu , Xiaolong Zhang , Tong Sha , Yankai Su , Wenming Zhao , Yiming Bao , Yongbiao Xue , Hua Chen","doi":"10.1016/j.gpb.2022.05.009","DOIUrl":null,"url":null,"abstract":"<div><p><strong>SARS-CoV-2</strong> is a new RNA virus affecting humans and spreads extensively throughout the world since its first outbreak in December, 2019. Whether the transmissibility and pathogenicity of SARS-CoV-2 in humans after zoonotic transfer are actively evolving, and driven by adaptation to the new host and environments is still under debate. Understanding the evolutionary mechanism underlying epidemiological and pathological characteristics of <strong>COVID-19</strong> is essential for predicting the epidemic trend, and providing guidance for disease control and treatments. Interrogating novel strategies for identifying <strong>natural selection</strong> using within-species polymorphisms and 3,674,076 SARS-CoV-2 genome sequences of 169 countries as of December 30, 2021, we demonstrate with population genetic evidence that during the course of SARS-CoV-2 pandemic in humans, 1) SARS-CoV-2 genomes are overall conserved under purifying selection, especially for the 14 genes related to viral RNA replication, transcription, and assembly; 2) ongoing positive selection is actively driving the evolution of 6 genes (<em>e.g.</em>, <em>S</em>, <em>ORF3a</em>, and <em>N</em>) that play critical roles in molecular processes involving pathogen–host interactions, including viral invasion into and egress from host cells, and viral inhibition and evasion of host immune response, possibly leading to high transmissibility and mild symptom in SARS-CoV-2 evolution. According to an established haplotype phylogenetic relationship of 138 viral clusters, a spatial and temporal landscape of 556 critical mutations is constructed based on their divergence among viral haplotype clusters or repeatedly increase in frequency within at least 2 clusters, of which multiple mutations potentially conferring alterations in viral transmissibility, pathogenicity, and virulence of SARS-CoV-2 are highlighted, warranting attention.</p></div>","PeriodicalId":12528,"journal":{"name":"Genomics, Proteomics & Bioinformatics","volume":"20 6","pages":"Pages 1214-1223"},"PeriodicalIF":7.9000,"publicationDate":"2022-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9233880/pdf/","citationCount":"10","resultStr":"{\"title\":\"Ongoing Positive Selection Drives the Evolution of SARS-CoV-2 Genomes\",\"authors\":\"Yali Hou , Shilei Zhao , Qi Liu , Xiaolong Zhang , Tong Sha , Yankai Su , Wenming Zhao , Yiming Bao , Yongbiao Xue , Hua Chen\",\"doi\":\"10.1016/j.gpb.2022.05.009\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p><strong>SARS-CoV-2</strong> is a new RNA virus affecting humans and spreads extensively throughout the world since its first outbreak in December, 2019. Whether the transmissibility and pathogenicity of SARS-CoV-2 in humans after zoonotic transfer are actively evolving, and driven by adaptation to the new host and environments is still under debate. Understanding the evolutionary mechanism underlying epidemiological and pathological characteristics of <strong>COVID-19</strong> is essential for predicting the epidemic trend, and providing guidance for disease control and treatments. Interrogating novel strategies for identifying <strong>natural selection</strong> using within-species polymorphisms and 3,674,076 SARS-CoV-2 genome sequences of 169 countries as of December 30, 2021, we demonstrate with population genetic evidence that during the course of SARS-CoV-2 pandemic in humans, 1) SARS-CoV-2 genomes are overall conserved under purifying selection, especially for the 14 genes related to viral RNA replication, transcription, and assembly; 2) ongoing positive selection is actively driving the evolution of 6 genes (<em>e.g.</em>, <em>S</em>, <em>ORF3a</em>, and <em>N</em>) that play critical roles in molecular processes involving pathogen–host interactions, including viral invasion into and egress from host cells, and viral inhibition and evasion of host immune response, possibly leading to high transmissibility and mild symptom in SARS-CoV-2 evolution. According to an established haplotype phylogenetic relationship of 138 viral clusters, a spatial and temporal landscape of 556 critical mutations is constructed based on their divergence among viral haplotype clusters or repeatedly increase in frequency within at least 2 clusters, of which multiple mutations potentially conferring alterations in viral transmissibility, pathogenicity, and virulence of SARS-CoV-2 are highlighted, warranting attention.</p></div>\",\"PeriodicalId\":12528,\"journal\":{\"name\":\"Genomics, Proteomics & Bioinformatics\",\"volume\":\"20 6\",\"pages\":\"Pages 1214-1223\"},\"PeriodicalIF\":7.9000,\"publicationDate\":\"2022-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9233880/pdf/\",\"citationCount\":\"10\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genomics, Proteomics & Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1672022922000791\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/6/26 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genomics, Proteomics & Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1672022922000791","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/6/26 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Ongoing Positive Selection Drives the Evolution of SARS-CoV-2 Genomes
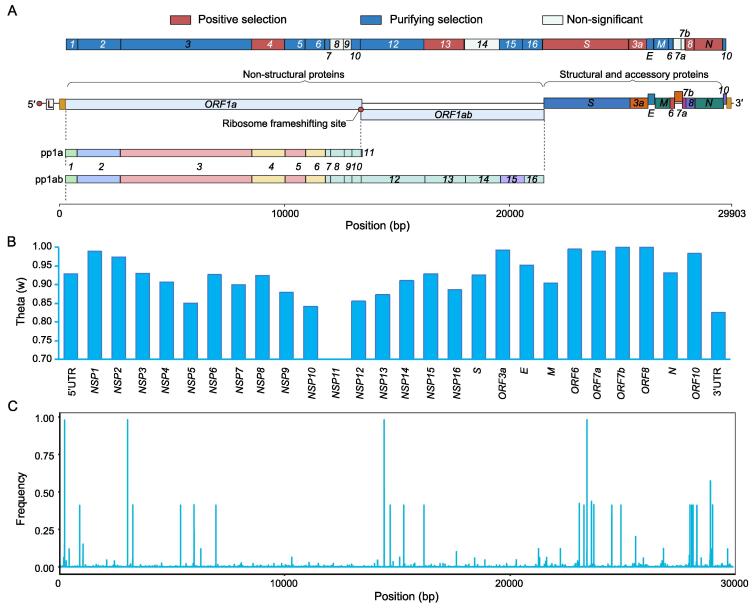
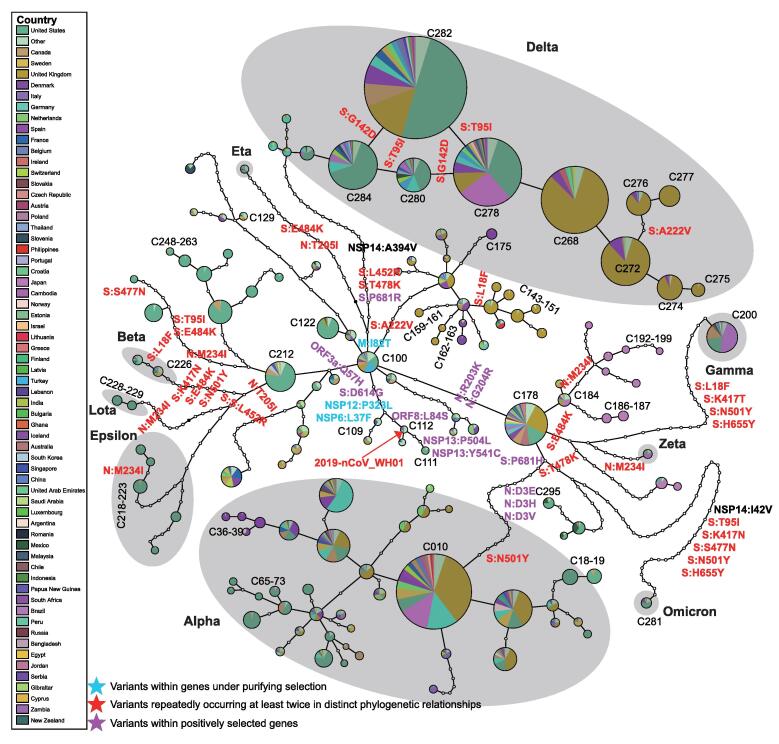
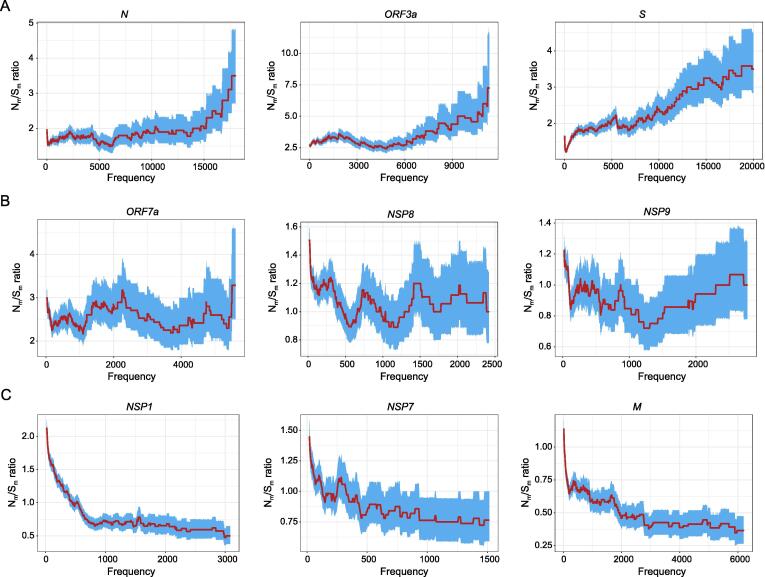
SARS-CoV-2 is a new RNA virus affecting humans and spreads extensively throughout the world since its first outbreak in December, 2019. Whether the transmissibility and pathogenicity of SARS-CoV-2 in humans after zoonotic transfer are actively evolving, and driven by adaptation to the new host and environments is still under debate. Understanding the evolutionary mechanism underlying epidemiological and pathological characteristics of COVID-19 is essential for predicting the epidemic trend, and providing guidance for disease control and treatments. Interrogating novel strategies for identifying natural selection using within-species polymorphisms and 3,674,076 SARS-CoV-2 genome sequences of 169 countries as of December 30, 2021, we demonstrate with population genetic evidence that during the course of SARS-CoV-2 pandemic in humans, 1) SARS-CoV-2 genomes are overall conserved under purifying selection, especially for the 14 genes related to viral RNA replication, transcription, and assembly; 2) ongoing positive selection is actively driving the evolution of 6 genes (e.g., S, ORF3a, and N) that play critical roles in molecular processes involving pathogen–host interactions, including viral invasion into and egress from host cells, and viral inhibition and evasion of host immune response, possibly leading to high transmissibility and mild symptom in SARS-CoV-2 evolution. According to an established haplotype phylogenetic relationship of 138 viral clusters, a spatial and temporal landscape of 556 critical mutations is constructed based on their divergence among viral haplotype clusters or repeatedly increase in frequency within at least 2 clusters, of which multiple mutations potentially conferring alterations in viral transmissibility, pathogenicity, and virulence of SARS-CoV-2 are highlighted, warranting attention.
期刊介绍:
Genomics, Proteomics and Bioinformatics (GPB) is the official journal of the Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation and Genetics Society of China. It aims to disseminate new developments in the field of omics and bioinformatics, publish high-quality discoveries quickly, and promote open access and online publication. GPB welcomes submissions in all areas of life science, biology, and biomedicine, with a focus on large data acquisition, analysis, and curation. Manuscripts covering omics and related bioinformatics topics are particularly encouraged. GPB is indexed/abstracted by PubMed/MEDLINE, PubMed Central, Scopus, BIOSIS Previews, Chemical Abstracts, CSCD, among others.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
