{"title":"激活 DRG 中的神经元和卫星神经胶质细胞可产生吗啡诱导的痛觉减退。","authors":"Shunsuke Yamakita, Daisuke Fujita, Kazuki Sudo, Daiki Ishikawa, Kohsuke Kushimoto, Yasuhiko Horii, Fumimasa Amaya","doi":"10.1177/17448069231181973","DOIUrl":null,"url":null,"abstract":"<p><p>Activation of neurons and glial cells in the dorsal root ganglion is one of the key mechanisms for the development of hyperalgesia. The aim of the present study was to examine the role of neuroglial activity in the development of opioid-induced hyperalgesia. Male rats were treated with morphine daily for 3 days. The resultant phosphorylation of extracellular signal-regulated kinase (ERK) 1/2 in the dorsal root ganglion was analyzed by immunohistochemistry and Western blotting. Pain hypersensitivity was analyzed using behavioral studies. The amount of cytokine expression in the dorsal root ganglion was also analyzed. Repeated morphine treatment induced hyperalgesia and marked induction of phosphorylated ERK1/2 in the neurons and satellite glial cells on day 3. An opioid receptor antagonist, toll like receptor-4 inhibitor, MAP/ERK kinase (MEK) inhibitor and gap junction inhibitor inhibited morphine-induced hyperalgesia and ERK1/2 phosphorylation. Morphine treatment induced alteration of cytokine expression, which was inhibited by the opioid receptor antagonist, toll like receptor-4 inhibitor, MEK inhibitor and gap junction inhibitor. Dexamethasone inhibited morphine-induced hyperalgesia and ERK1/2 phosphorylation after morphine treatment. The peripherally restricted opioid receptor antagonist, methylnaltrexone, inhibited hyperalgesia and ERK1/2 phosphorylation. Morphine activates ERK1/2 in neurons and satellite glial cells in the dorsal root ganglion via the opioid receptor and toll like receptor-4. ERK1/2 phosphorylation is gap junction-dependent and is associated with the alteration of cytokine expression. Inhibition of neuroinflammation by activation of neurons and glia might be a promising target to prevent opioid-induced hyperalgesia.</p>","PeriodicalId":19010,"journal":{"name":"Molecular Pain","volume":" ","pages":"17448069231181973"},"PeriodicalIF":2.8000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/17/4c/10.1177_17448069231181973.PMC10291868.pdf","citationCount":"0","resultStr":"{\"title\":\"Activation of neurons and satellite glial cells in the DRG produces morphine-induced hyperalgesia.\",\"authors\":\"Shunsuke Yamakita, Daisuke Fujita, Kazuki Sudo, Daiki Ishikawa, Kohsuke Kushimoto, Yasuhiko Horii, Fumimasa Amaya\",\"doi\":\"10.1177/17448069231181973\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Activation of neurons and glial cells in the dorsal root ganglion is one of the key mechanisms for the development of hyperalgesia. The aim of the present study was to examine the role of neuroglial activity in the development of opioid-induced hyperalgesia. Male rats were treated with morphine daily for 3 days. The resultant phosphorylation of extracellular signal-regulated kinase (ERK) 1/2 in the dorsal root ganglion was analyzed by immunohistochemistry and Western blotting. Pain hypersensitivity was analyzed using behavioral studies. The amount of cytokine expression in the dorsal root ganglion was also analyzed. Repeated morphine treatment induced hyperalgesia and marked induction of phosphorylated ERK1/2 in the neurons and satellite glial cells on day 3. An opioid receptor antagonist, toll like receptor-4 inhibitor, MAP/ERK kinase (MEK) inhibitor and gap junction inhibitor inhibited morphine-induced hyperalgesia and ERK1/2 phosphorylation. Morphine treatment induced alteration of cytokine expression, which was inhibited by the opioid receptor antagonist, toll like receptor-4 inhibitor, MEK inhibitor and gap junction inhibitor. Dexamethasone inhibited morphine-induced hyperalgesia and ERK1/2 phosphorylation after morphine treatment. The peripherally restricted opioid receptor antagonist, methylnaltrexone, inhibited hyperalgesia and ERK1/2 phosphorylation. Morphine activates ERK1/2 in neurons and satellite glial cells in the dorsal root ganglion via the opioid receptor and toll like receptor-4. ERK1/2 phosphorylation is gap junction-dependent and is associated with the alteration of cytokine expression. Inhibition of neuroinflammation by activation of neurons and glia might be a promising target to prevent opioid-induced hyperalgesia.</p>\",\"PeriodicalId\":19010,\"journal\":{\"name\":\"Molecular Pain\",\"volume\":\" \",\"pages\":\"17448069231181973\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/17/4c/10.1177_17448069231181973.PMC10291868.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Pain\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1177/17448069231181973\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"NEUROSCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Pain","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1177/17448069231181973","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 0
摘要
激活背根神经节中的神经元和神经胶质细胞是产生超痛感的关键机制之一。本研究旨在探讨神经胶质细胞活动在阿片类药物诱导的痛觉减退中的作用。雄性大鼠每天接受吗啡治疗 3 天。通过免疫组织化学和 Western 印迹法分析了背根神经节中细胞外信号调节激酶(ERK)1/2 的磷酸化情况。通过行为研究分析了痛觉过敏性。还分析了背根神经节中细胞因子的表达量。重复吗啡处理可诱导痛觉减退,并在第3天明显诱导神经元和卫星神经胶质细胞磷酸化ERK1/2。阿片受体拮抗剂、类收费受体-4抑制剂、MAP/ERK激酶(MEK)抑制剂和间隙连接抑制剂抑制了吗啡诱导的痛觉减退和ERK1/2磷酸化。阿片受体拮抗剂、收费样受体-4 抑制剂、MEK 抑制剂和间隙连接抑制剂可抑制吗啡诱导的细胞因子表达。地塞米松可抑制吗啡诱导的痛觉减退和吗啡治疗后的ERK1/2磷酸化。外周限制性阿片受体拮抗剂甲纳曲酮抑制了超痛感和ERK1/2磷酸化。吗啡通过阿片受体和类收费受体-4激活背根神经节神经元和卫星胶质细胞中的ERK1/2。ERK1/2 磷酸化依赖于间隙连接,并与细胞因子表达的改变有关。通过激活神经元和神经胶质细胞来抑制神经炎症可能是预防阿片类药物引起的痛觉减退的一个有希望的靶点。
Activation of neurons and satellite glial cells in the DRG produces morphine-induced hyperalgesia.
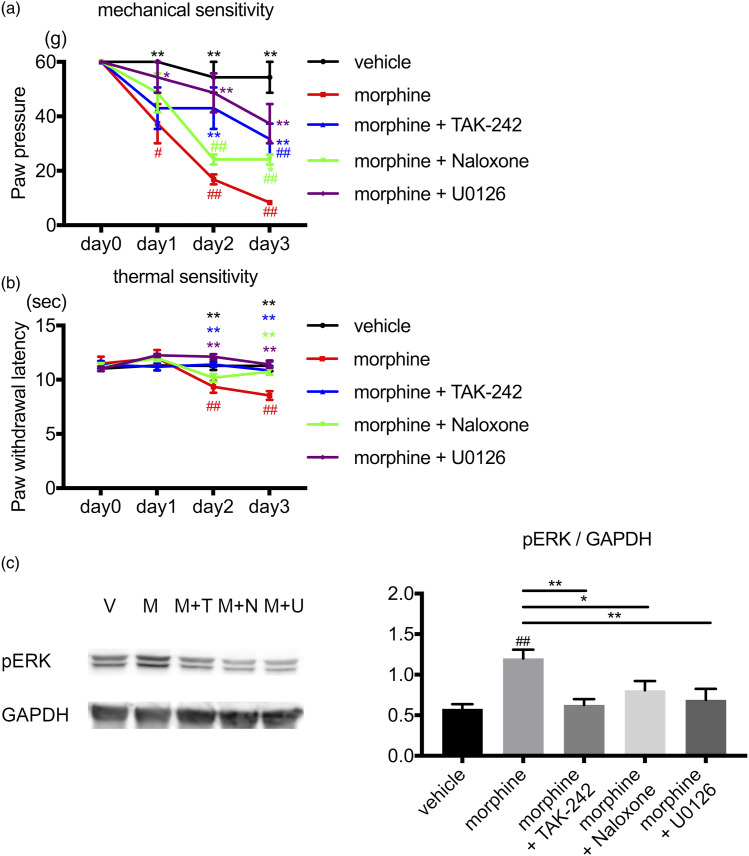
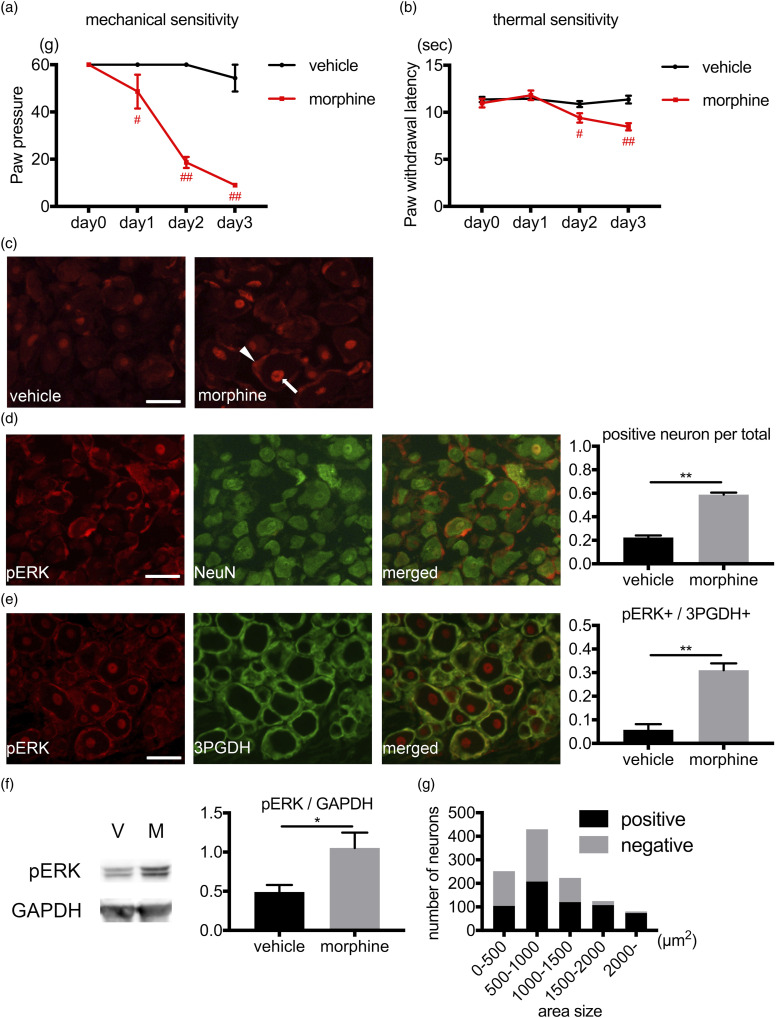
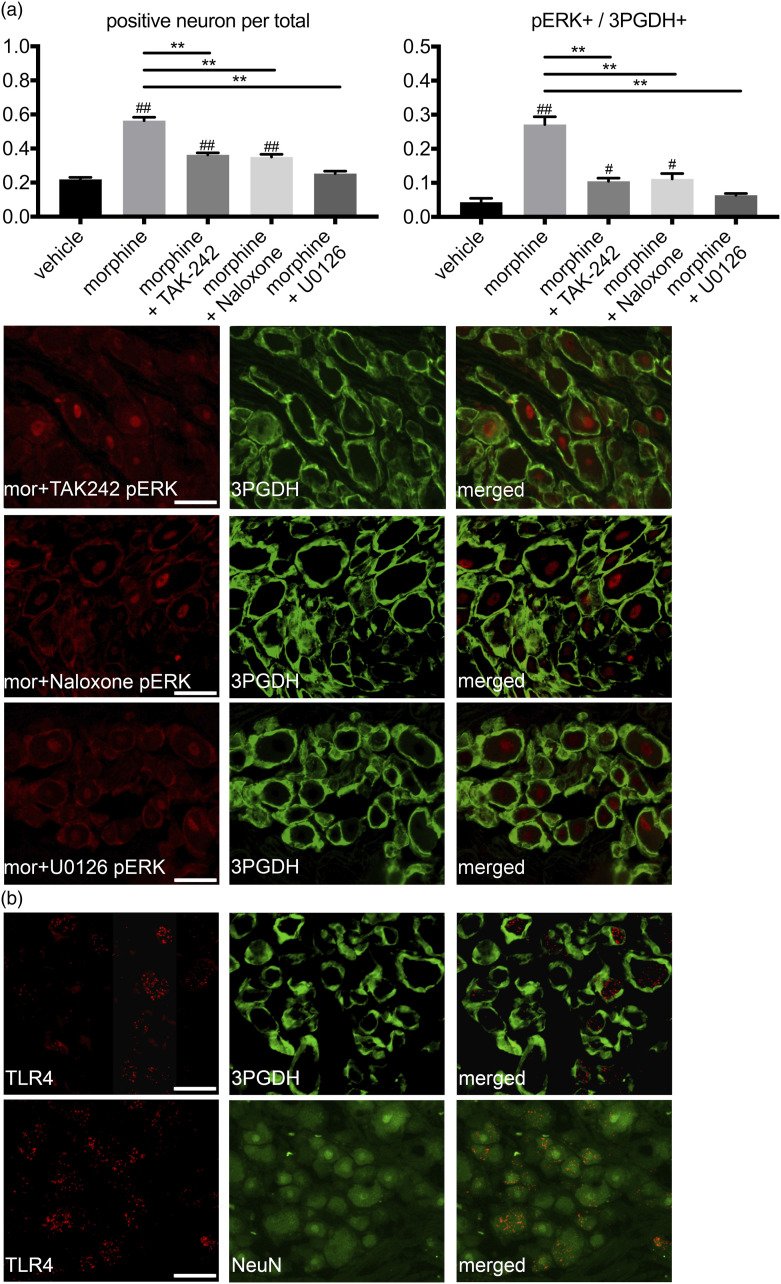
Activation of neurons and glial cells in the dorsal root ganglion is one of the key mechanisms for the development of hyperalgesia. The aim of the present study was to examine the role of neuroglial activity in the development of opioid-induced hyperalgesia. Male rats were treated with morphine daily for 3 days. The resultant phosphorylation of extracellular signal-regulated kinase (ERK) 1/2 in the dorsal root ganglion was analyzed by immunohistochemistry and Western blotting. Pain hypersensitivity was analyzed using behavioral studies. The amount of cytokine expression in the dorsal root ganglion was also analyzed. Repeated morphine treatment induced hyperalgesia and marked induction of phosphorylated ERK1/2 in the neurons and satellite glial cells on day 3. An opioid receptor antagonist, toll like receptor-4 inhibitor, MAP/ERK kinase (MEK) inhibitor and gap junction inhibitor inhibited morphine-induced hyperalgesia and ERK1/2 phosphorylation. Morphine treatment induced alteration of cytokine expression, which was inhibited by the opioid receptor antagonist, toll like receptor-4 inhibitor, MEK inhibitor and gap junction inhibitor. Dexamethasone inhibited morphine-induced hyperalgesia and ERK1/2 phosphorylation after morphine treatment. The peripherally restricted opioid receptor antagonist, methylnaltrexone, inhibited hyperalgesia and ERK1/2 phosphorylation. Morphine activates ERK1/2 in neurons and satellite glial cells in the dorsal root ganglion via the opioid receptor and toll like receptor-4. ERK1/2 phosphorylation is gap junction-dependent and is associated with the alteration of cytokine expression. Inhibition of neuroinflammation by activation of neurons and glia might be a promising target to prevent opioid-induced hyperalgesia.
期刊介绍:
Molecular Pain is a peer-reviewed, open access journal that considers manuscripts in pain research at the cellular, subcellular and molecular levels. Molecular Pain provides a forum for molecular pain scientists to communicate their research findings in a targeted manner to others in this important and growing field.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
