{"title":"比较力场研究含锌指蛋白NPL4,二硫仑在癌症治疗中的靶点","authors":"Simone Scrima , Matteo Tiberti , Ulf Ryde , Matteo Lambrughi , Elena Papaleo","doi":"10.1016/j.bbapap.2023.140921","DOIUrl":null,"url":null,"abstract":"<div><p>Molecular dynamics (MD) simulations are a powerful approach to studying the structure and dynamics of proteins related to health and disease. Advances in the MD field allow modeling proteins with high accuracy. However, modeling metal ions and their interactions with proteins is still challenging. NPL4 is a zinc-binding protein and works as a cofactor for p97 to regulate protein homeostasis. NPL4 is of biomedical importance and has been proposed as the target of disulfiram, a drug recently repurposed for cancer treatment. Experimental studies proposed that the disulfiram metabolites, bis-(diethyldithiocarbamate)‑copper and cupric ions, induce NPL4 misfolding and aggregation. However, the molecular details of their interactions with NPL4 and consequent structural effects are still elusive. Here, biomolecular simulations can help to shed light on the related structural details. To apply MD simulations to NPL4 and its interaction with copper the first important step is identifying a suitable force field to describe the protein in its zinc-bound states. We examined different sets of non-bonded parameters because we want to study the misfolding mechanism and cannot rule out that the zinc may detach from the protein during the process and copper replaces it. We investigated the force-field ability to model the coordination geometry of the metal ions by comparing the results from MD simulations with optimized geometries from quantum mechanics (QM) calculations using model systems of NPL4. Furthermore, we investigated the performance of a force field including bonded parameters to treat copper ions in NPL4 that we obtained based on QM calculations.</p></div>","PeriodicalId":8760,"journal":{"name":"Biochimica et biophysica acta. Proteins and proteomics","volume":"1871 4","pages":"Article 140921"},"PeriodicalIF":2.3000,"publicationDate":"2023-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":"{\"title\":\"Comparison of force fields to study the zinc-finger containing protein NPL4, a target for disulfiram in cancer therapy\",\"authors\":\"Simone Scrima , Matteo Tiberti , Ulf Ryde , Matteo Lambrughi , Elena Papaleo\",\"doi\":\"10.1016/j.bbapap.2023.140921\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Molecular dynamics (MD) simulations are a powerful approach to studying the structure and dynamics of proteins related to health and disease. Advances in the MD field allow modeling proteins with high accuracy. However, modeling metal ions and their interactions with proteins is still challenging. NPL4 is a zinc-binding protein and works as a cofactor for p97 to regulate protein homeostasis. NPL4 is of biomedical importance and has been proposed as the target of disulfiram, a drug recently repurposed for cancer treatment. Experimental studies proposed that the disulfiram metabolites, bis-(diethyldithiocarbamate)‑copper and cupric ions, induce NPL4 misfolding and aggregation. However, the molecular details of their interactions with NPL4 and consequent structural effects are still elusive. Here, biomolecular simulations can help to shed light on the related structural details. To apply MD simulations to NPL4 and its interaction with copper the first important step is identifying a suitable force field to describe the protein in its zinc-bound states. We examined different sets of non-bonded parameters because we want to study the misfolding mechanism and cannot rule out that the zinc may detach from the protein during the process and copper replaces it. We investigated the force-field ability to model the coordination geometry of the metal ions by comparing the results from MD simulations with optimized geometries from quantum mechanics (QM) calculations using model systems of NPL4. Furthermore, we investigated the performance of a force field including bonded parameters to treat copper ions in NPL4 that we obtained based on QM calculations.</p></div>\",\"PeriodicalId\":8760,\"journal\":{\"name\":\"Biochimica et biophysica acta. Proteins and proteomics\",\"volume\":\"1871 4\",\"pages\":\"Article 140921\"},\"PeriodicalIF\":2.3000,\"publicationDate\":\"2023-07-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biochimica et biophysica acta. Proteins and proteomics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1570963923000353\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/5/24 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biochimica et biophysica acta. Proteins and proteomics","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1570963923000353","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/5/24 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Comparison of force fields to study the zinc-finger containing protein NPL4, a target for disulfiram in cancer therapy
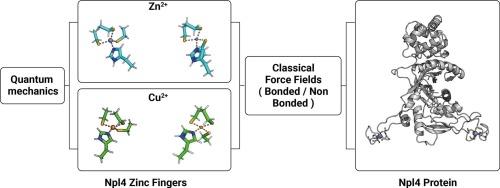
Molecular dynamics (MD) simulations are a powerful approach to studying the structure and dynamics of proteins related to health and disease. Advances in the MD field allow modeling proteins with high accuracy. However, modeling metal ions and their interactions with proteins is still challenging. NPL4 is a zinc-binding protein and works as a cofactor for p97 to regulate protein homeostasis. NPL4 is of biomedical importance and has been proposed as the target of disulfiram, a drug recently repurposed for cancer treatment. Experimental studies proposed that the disulfiram metabolites, bis-(diethyldithiocarbamate)‑copper and cupric ions, induce NPL4 misfolding and aggregation. However, the molecular details of their interactions with NPL4 and consequent structural effects are still elusive. Here, biomolecular simulations can help to shed light on the related structural details. To apply MD simulations to NPL4 and its interaction with copper the first important step is identifying a suitable force field to describe the protein in its zinc-bound states. We examined different sets of non-bonded parameters because we want to study the misfolding mechanism and cannot rule out that the zinc may detach from the protein during the process and copper replaces it. We investigated the force-field ability to model the coordination geometry of the metal ions by comparing the results from MD simulations with optimized geometries from quantum mechanics (QM) calculations using model systems of NPL4. Furthermore, we investigated the performance of a force field including bonded parameters to treat copper ions in NPL4 that we obtained based on QM calculations.
期刊介绍:
BBA Proteins and Proteomics covers protein structure conformation and dynamics; protein folding; protein-ligand interactions; enzyme mechanisms, models and kinetics; protein physical properties and spectroscopy; and proteomics and bioinformatics analyses of protein structure, protein function, or protein regulation.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
