Shiroye Olukayode, Charlotte Froese Fischer, Anatoliy Volkov
{"title":"重访相对论狄拉克-哈特里-福克x射线散射因子。2Z = 3-112原子的化学相关阳离子和选定的一价阴离子。","authors":"Shiroye Olukayode, Charlotte Froese Fischer, Anatoliy Volkov","doi":"10.1107/S205327332300116X","DOIUrl":null,"url":null,"abstract":"<p><p>The previously described approach for determination of the relativistic atomic X-ray scattering factors (XRSFs) at the Dirac-Hartree-Fock level [Olukayode et al. (2023). Acta Cryst. A79, 59-79] has been used to evaluate the XRSFs for a total of 318 species including all chemically relevant cations [Greenwood & Earnshaw (1997). Chemistry of the Elements], six monovalent anions (O<sup>-</sup>, F<sup>-</sup>, Cl<sup>-</sup>, Br<sup>-</sup>, I<sup>-</sup>, At<sup>-</sup>), the ns<sup>1</sup>np<sup>3</sup> excited (valence) states of carbon and silicon, and several exotic cations (Db<sup>5+</sup>, Sg<sup>6+</sup>, Bh<sup>7+</sup>, Hs<sup>8+</sup> and Cn<sup>2+</sup>) for which the chemical compounds have been recently identified, thus significantly extending the coverage relative to all the earlier studies. Unlike the data currently recommended by the International Union of Crystallography (IUCr) [Maslen et al. (2006). International Tables for Crystallography, Vol. C, Section 6.1.1, pp. 554-589], which originate from different levels of theory including the non-relativistic Hartree-Fock and correlated methods, as well as the relativistic Dirac-Slater calculations, the re-determined XRSFs come from a uniform treatment of all species within the same relativistic B-spline Dirac-Hartree-Fock approach [Zatsarinny & Froese Fischer (2016). Comput. Phys. Comm. 202, 287-303] that includes the Breit interaction correction and the Fermi nuclear charge density model. While it was not possible to compare the quality of the generated wavefunctions with that from the previous studies due to a lack (to the best of our knowledge) of such data in the literature, a careful comparison of the total electronic energies and the estimated atomic ionization energies with experimental and theoretical values from other studies instils confidence in the quality of the calculations. A combination of the B-spline approach and a fine radial grid allowed for a precise determination of the XRSFs for each species in the entire 0 ≤ sin θ/λ ≤ 6 Å<sup>-1</sup> range, thus avoiding the necessity for extrapolation in the 2 ≤ sin θ/λ ≤ 6 Å<sup>-1</sup> interval which, as was shown in the first study, may lead to inconsistencies. In contrast to the Rez et al. work [Acta Cryst. (1994), A50, 481-497], no additional approximations were introduced when calculating wavefunctions for the anions. The conventional and extended expansions were employed to produce interpolating functions for each species in both the 0 ≤ sin θ/λ ≤ 2 Å<sup>-1</sup> and 2 ≤ sin θ/λ ≤ 6 Å<sup>-1</sup> intervals, with the extended expansions offering a significantly better accuracy at a minimal computational overhead. The combined results of this and the previous study may be used to update the XRSFs for neutral atoms and ions listed in Vol. C of the 2006 edition of International Tables for Crystallography.</p>","PeriodicalId":106,"journal":{"name":"Acta Crystallographica Section A: Foundations and Advances","volume":"79 Pt 3","pages":"229-245"},"PeriodicalIF":1.8000,"publicationDate":"2023-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Revisited relativistic Dirac-Hartree-Fock X-ray scattering factors. II. Chemically relevant cations and selected monovalent anions for atoms with Z = 3-112.\",\"authors\":\"Shiroye Olukayode, Charlotte Froese Fischer, Anatoliy Volkov\",\"doi\":\"10.1107/S205327332300116X\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The previously described approach for determination of the relativistic atomic X-ray scattering factors (XRSFs) at the Dirac-Hartree-Fock level [Olukayode et al. (2023). Acta Cryst. A79, 59-79] has been used to evaluate the XRSFs for a total of 318 species including all chemically relevant cations [Greenwood & Earnshaw (1997). Chemistry of the Elements], six monovalent anions (O<sup>-</sup>, F<sup>-</sup>, Cl<sup>-</sup>, Br<sup>-</sup>, I<sup>-</sup>, At<sup>-</sup>), the ns<sup>1</sup>np<sup>3</sup> excited (valence) states of carbon and silicon, and several exotic cations (Db<sup>5+</sup>, Sg<sup>6+</sup>, Bh<sup>7+</sup>, Hs<sup>8+</sup> and Cn<sup>2+</sup>) for which the chemical compounds have been recently identified, thus significantly extending the coverage relative to all the earlier studies. Unlike the data currently recommended by the International Union of Crystallography (IUCr) [Maslen et al. (2006). International Tables for Crystallography, Vol. C, Section 6.1.1, pp. 554-589], which originate from different levels of theory including the non-relativistic Hartree-Fock and correlated methods, as well as the relativistic Dirac-Slater calculations, the re-determined XRSFs come from a uniform treatment of all species within the same relativistic B-spline Dirac-Hartree-Fock approach [Zatsarinny & Froese Fischer (2016). Comput. Phys. Comm. 202, 287-303] that includes the Breit interaction correction and the Fermi nuclear charge density model. While it was not possible to compare the quality of the generated wavefunctions with that from the previous studies due to a lack (to the best of our knowledge) of such data in the literature, a careful comparison of the total electronic energies and the estimated atomic ionization energies with experimental and theoretical values from other studies instils confidence in the quality of the calculations. A combination of the B-spline approach and a fine radial grid allowed for a precise determination of the XRSFs for each species in the entire 0 ≤ sin θ/λ ≤ 6 Å<sup>-1</sup> range, thus avoiding the necessity for extrapolation in the 2 ≤ sin θ/λ ≤ 6 Å<sup>-1</sup> interval which, as was shown in the first study, may lead to inconsistencies. In contrast to the Rez et al. work [Acta Cryst. (1994), A50, 481-497], no additional approximations were introduced when calculating wavefunctions for the anions. The conventional and extended expansions were employed to produce interpolating functions for each species in both the 0 ≤ sin θ/λ ≤ 2 Å<sup>-1</sup> and 2 ≤ sin θ/λ ≤ 6 Å<sup>-1</sup> intervals, with the extended expansions offering a significantly better accuracy at a minimal computational overhead. The combined results of this and the previous study may be used to update the XRSFs for neutral atoms and ions listed in Vol. C of the 2006 edition of International Tables for Crystallography.</p>\",\"PeriodicalId\":106,\"journal\":{\"name\":\"Acta Crystallographica Section A: Foundations and Advances\",\"volume\":\"79 Pt 3\",\"pages\":\"229-245\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2023-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Acta Crystallographica Section A: Foundations and Advances\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://doi.org/10.1107/S205327332300116X\",\"RegionNum\":4,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Crystallographica Section A: Foundations and Advances","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1107/S205327332300116X","RegionNum":4,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Revisited relativistic Dirac-Hartree-Fock X-ray scattering factors. II. Chemically relevant cations and selected monovalent anions for atoms with Z = 3-112.
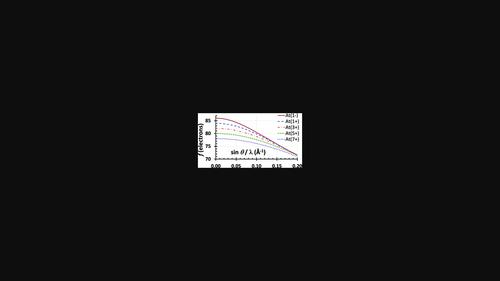
The previously described approach for determination of the relativistic atomic X-ray scattering factors (XRSFs) at the Dirac-Hartree-Fock level [Olukayode et al. (2023). Acta Cryst. A79, 59-79] has been used to evaluate the XRSFs for a total of 318 species including all chemically relevant cations [Greenwood & Earnshaw (1997). Chemistry of the Elements], six monovalent anions (O-, F-, Cl-, Br-, I-, At-), the ns1np3 excited (valence) states of carbon and silicon, and several exotic cations (Db5+, Sg6+, Bh7+, Hs8+ and Cn2+) for which the chemical compounds have been recently identified, thus significantly extending the coverage relative to all the earlier studies. Unlike the data currently recommended by the International Union of Crystallography (IUCr) [Maslen et al. (2006). International Tables for Crystallography, Vol. C, Section 6.1.1, pp. 554-589], which originate from different levels of theory including the non-relativistic Hartree-Fock and correlated methods, as well as the relativistic Dirac-Slater calculations, the re-determined XRSFs come from a uniform treatment of all species within the same relativistic B-spline Dirac-Hartree-Fock approach [Zatsarinny & Froese Fischer (2016). Comput. Phys. Comm. 202, 287-303] that includes the Breit interaction correction and the Fermi nuclear charge density model. While it was not possible to compare the quality of the generated wavefunctions with that from the previous studies due to a lack (to the best of our knowledge) of such data in the literature, a careful comparison of the total electronic energies and the estimated atomic ionization energies with experimental and theoretical values from other studies instils confidence in the quality of the calculations. A combination of the B-spline approach and a fine radial grid allowed for a precise determination of the XRSFs for each species in the entire 0 ≤ sin θ/λ ≤ 6 Å-1 range, thus avoiding the necessity for extrapolation in the 2 ≤ sin θ/λ ≤ 6 Å-1 interval which, as was shown in the first study, may lead to inconsistencies. In contrast to the Rez et al. work [Acta Cryst. (1994), A50, 481-497], no additional approximations were introduced when calculating wavefunctions for the anions. The conventional and extended expansions were employed to produce interpolating functions for each species in both the 0 ≤ sin θ/λ ≤ 2 Å-1 and 2 ≤ sin θ/λ ≤ 6 Å-1 intervals, with the extended expansions offering a significantly better accuracy at a minimal computational overhead. The combined results of this and the previous study may be used to update the XRSFs for neutral atoms and ions listed in Vol. C of the 2006 edition of International Tables for Crystallography.
期刊介绍:
Acta Crystallographica Section A: Foundations and Advances publishes articles reporting advances in the theory and practice of all areas of crystallography in the broadest sense. As well as traditional crystallography, this includes nanocrystals, metacrystals, amorphous materials, quasicrystals, synchrotron and XFEL studies, coherent scattering, diffraction imaging, time-resolved studies and the structure of strain and defects in materials.
The journal has two parts, a rapid-publication Advances section and the traditional Foundations section. Articles for the Advances section are of particularly high value and impact. They receive expedited treatment and may be highlighted by an accompanying scientific commentary article and a press release. Further details are given in the November 2013 Editorial.
The central themes of the journal are, on the one hand, experimental and theoretical studies of the properties and arrangements of atoms, ions and molecules in condensed matter, periodic, quasiperiodic or amorphous, ideal or real, and, on the other, the theoretical and experimental aspects of the various methods to determine these properties and arrangements.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
