Renfei Ma , Shangfu Li , Wenshuo Li , Lantian Yao , Hsien-Da Huang , Tzong-Yi Lee
{"title":"KinasePhos 3.0:激酶特异性磷酸化位点预测的重新设计和扩展","authors":"Renfei Ma , Shangfu Li , Wenshuo Li , Lantian Yao , Hsien-Da Huang , Tzong-Yi Lee","doi":"10.1016/j.gpb.2022.06.004","DOIUrl":null,"url":null,"abstract":"<div><p>The purpose of this work is to enhance KinasePhos, a machine learning-based <strong>kinase-specific phosphorylation site prediction</strong> tool. Experimentally verified kinase-specific phosphorylation data were collected from PhosphoSitePlus, UniProtKB, the GPS 5.0, and Phospho.ELM. In total, 41,421 experimentally verified kinase-specific phosphorylation sites were identified. A total of 1380 unique kinases were identified, including 753 with existing classification information from KinBase and the remaining 627 annotated by building a phylogenetic tree. Based on this kinase classification, a total of 771 predictive models were built at the individual, family, and group levels, using at least 15 experimentally verified substrate sites in positive training datasets. The improved models demonstrated their effectiveness compared with other prediction tools. For example, the prediction of sites phosphorylated by the protein kinase B, casein kinase 2, and protein kinase A families had accuracies of 94.5%, 92.5%, and 90.0%, respectively. The average prediction accuracy for all 771 models was 87.2%. For enhancing interpretability, the SHapley Additive exPlanations (SHAP) method was employed to assess feature importance. The web interface of KinasePhos 3.0 has been redesigned to provide comprehensive annotations of kinase-specific phosphorylation sites on multiple proteins. Additionally, considering the large scale of phosphoproteomic data, a downloadable prediction tool is available at <span>https://awi.cuhk.edu.cn/KinasePhos/download.html</span><svg><path></path></svg> or <span>https://github.com/tom-209/KinasePhos-3.0-executable-file</span><svg><path></path></svg>.</p></div>","PeriodicalId":12528,"journal":{"name":"Genomics, Proteomics & Bioinformatics","volume":"21 1","pages":"Pages 228-241"},"PeriodicalIF":7.9000,"publicationDate":"2023-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10373160/pdf/","citationCount":"11","resultStr":"{\"title\":\"KinasePhos 3.0: Redesign and Expansion of the Prediction on Kinase-specific Phosphorylation Sites\",\"authors\":\"Renfei Ma , Shangfu Li , Wenshuo Li , Lantian Yao , Hsien-Da Huang , Tzong-Yi Lee\",\"doi\":\"10.1016/j.gpb.2022.06.004\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The purpose of this work is to enhance KinasePhos, a machine learning-based <strong>kinase-specific phosphorylation site prediction</strong> tool. Experimentally verified kinase-specific phosphorylation data were collected from PhosphoSitePlus, UniProtKB, the GPS 5.0, and Phospho.ELM. In total, 41,421 experimentally verified kinase-specific phosphorylation sites were identified. A total of 1380 unique kinases were identified, including 753 with existing classification information from KinBase and the remaining 627 annotated by building a phylogenetic tree. Based on this kinase classification, a total of 771 predictive models were built at the individual, family, and group levels, using at least 15 experimentally verified substrate sites in positive training datasets. The improved models demonstrated their effectiveness compared with other prediction tools. For example, the prediction of sites phosphorylated by the protein kinase B, casein kinase 2, and protein kinase A families had accuracies of 94.5%, 92.5%, and 90.0%, respectively. The average prediction accuracy for all 771 models was 87.2%. For enhancing interpretability, the SHapley Additive exPlanations (SHAP) method was employed to assess feature importance. The web interface of KinasePhos 3.0 has been redesigned to provide comprehensive annotations of kinase-specific phosphorylation sites on multiple proteins. Additionally, considering the large scale of phosphoproteomic data, a downloadable prediction tool is available at <span>https://awi.cuhk.edu.cn/KinasePhos/download.html</span><svg><path></path></svg> or <span>https://github.com/tom-209/KinasePhos-3.0-executable-file</span><svg><path></path></svg>.</p></div>\",\"PeriodicalId\":12528,\"journal\":{\"name\":\"Genomics, Proteomics & Bioinformatics\",\"volume\":\"21 1\",\"pages\":\"Pages 228-241\"},\"PeriodicalIF\":7.9000,\"publicationDate\":\"2023-02-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10373160/pdf/\",\"citationCount\":\"11\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genomics, Proteomics & Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S167202292200081X\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/7/1 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genomics, Proteomics & Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S167202292200081X","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/7/1 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
KinasePhos 3.0: Redesign and Expansion of the Prediction on Kinase-specific Phosphorylation Sites
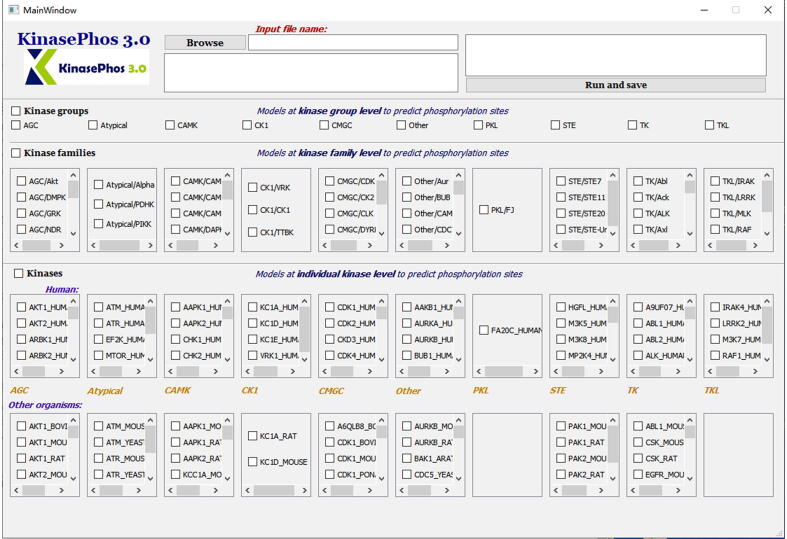
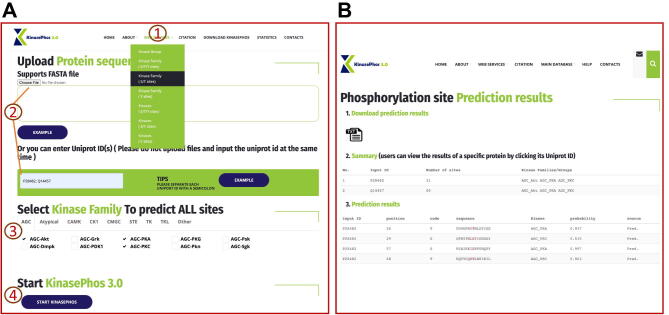
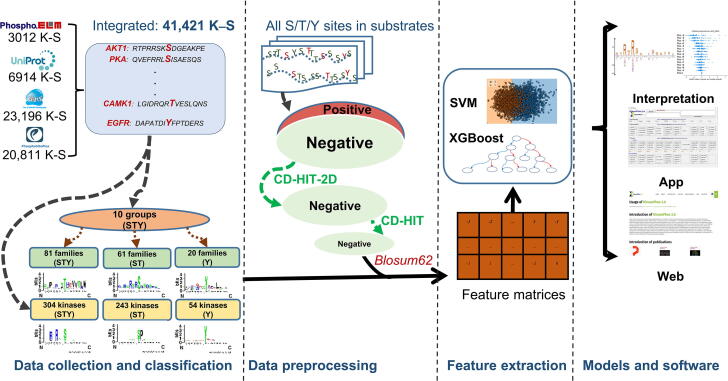
The purpose of this work is to enhance KinasePhos, a machine learning-based kinase-specific phosphorylation site prediction tool. Experimentally verified kinase-specific phosphorylation data were collected from PhosphoSitePlus, UniProtKB, the GPS 5.0, and Phospho.ELM. In total, 41,421 experimentally verified kinase-specific phosphorylation sites were identified. A total of 1380 unique kinases were identified, including 753 with existing classification information from KinBase and the remaining 627 annotated by building a phylogenetic tree. Based on this kinase classification, a total of 771 predictive models were built at the individual, family, and group levels, using at least 15 experimentally verified substrate sites in positive training datasets. The improved models demonstrated their effectiveness compared with other prediction tools. For example, the prediction of sites phosphorylated by the protein kinase B, casein kinase 2, and protein kinase A families had accuracies of 94.5%, 92.5%, and 90.0%, respectively. The average prediction accuracy for all 771 models was 87.2%. For enhancing interpretability, the SHapley Additive exPlanations (SHAP) method was employed to assess feature importance. The web interface of KinasePhos 3.0 has been redesigned to provide comprehensive annotations of kinase-specific phosphorylation sites on multiple proteins. Additionally, considering the large scale of phosphoproteomic data, a downloadable prediction tool is available at https://awi.cuhk.edu.cn/KinasePhos/download.html or https://github.com/tom-209/KinasePhos-3.0-executable-file.
期刊介绍:
Genomics, Proteomics and Bioinformatics (GPB) is the official journal of the Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation and Genetics Society of China. It aims to disseminate new developments in the field of omics and bioinformatics, publish high-quality discoveries quickly, and promote open access and online publication. GPB welcomes submissions in all areas of life science, biology, and biomedicine, with a focus on large data acquisition, analysis, and curation. Manuscripts covering omics and related bioinformatics topics are particularly encouraged. GPB is indexed/abstracted by PubMed/MEDLINE, PubMed Central, Scopus, BIOSIS Previews, Chemical Abstracts, CSCD, among others.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
