{"title":"DeepAutoGlioma: a deep learning autoencoder-based multi-omics data integration and classification tools for glioma subtyping.","authors":"Sana Munquad, Asim Bikas Das","doi":"10.1186/s13040-023-00349-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objective: </strong>The classification of glioma subtypes is essential for precision therapy. Due to the heterogeneity of gliomas, the subtype-specific molecular pattern can be captured by integrating and analyzing high-throughput omics data from different genomic layers. The development of a deep-learning framework enables the integration of multi-omics data to classify the glioma subtypes to support the clinical diagnosis.</p><p><strong>Results: </strong>Transcriptome and methylome data of glioma patients were preprocessed, and differentially expressed features from both datasets were identified. Subsequently, a Cox regression analysis determined genes and CpGs associated with survival. Gene set enrichment analysis was carried out to examine the biological significance of the features. Further, we identified CpG and gene pairs by mapping them in the promoter region of corresponding genes. The methylation and gene expression levels of these CpGs and genes were embedded in a lower-dimensional space with an autoencoder. Next, ANN and CNN were used to classify subtypes using the latent features from embedding space. CNN performs better than ANN for subtyping lower-grade gliomas (LGG) and glioblastoma multiforme (GBM). The subtyping accuracy of CNN was 98.03% (± 0.06) and 94.07% (± 0.01) in LGG and GBM, respectively. The precision of the models was 97.67% in LGG and 90.40% in GBM. The model sensitivity was 96.96% in LGG and 91.18% in GBM. Additionally, we observed the superior performance of CNN with external datasets. The genes and CpGs pairs used to develop the model showed better performance than the random CpGs-gene pairs, preprocessed data, and single omics data.</p><p><strong>Conclusions: </strong>The current study showed that a novel feature selection and data integration strategy led to the development of DeepAutoGlioma, an effective framework for diagnosing glioma subtypes.</p>","PeriodicalId":48947,"journal":{"name":"Biodata Mining","volume":"16 1","pages":"32"},"PeriodicalIF":6.1000,"publicationDate":"2023-11-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10652591/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biodata Mining","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13040-023-00349-7","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background and objective: The classification of glioma subtypes is essential for precision therapy. Due to the heterogeneity of gliomas, the subtype-specific molecular pattern can be captured by integrating and analyzing high-throughput omics data from different genomic layers. The development of a deep-learning framework enables the integration of multi-omics data to classify the glioma subtypes to support the clinical diagnosis.
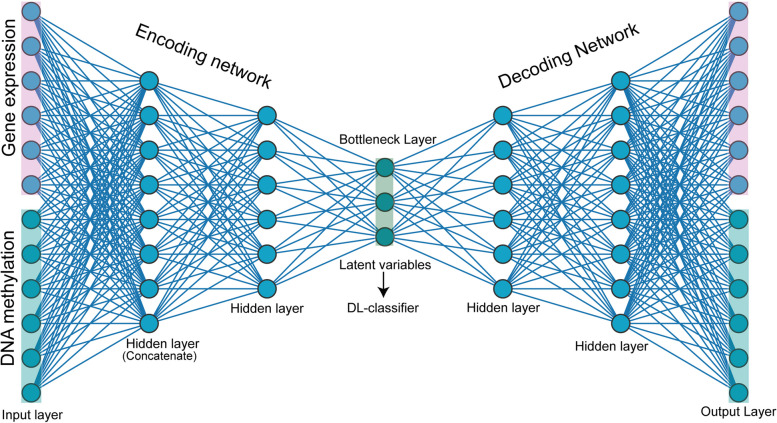
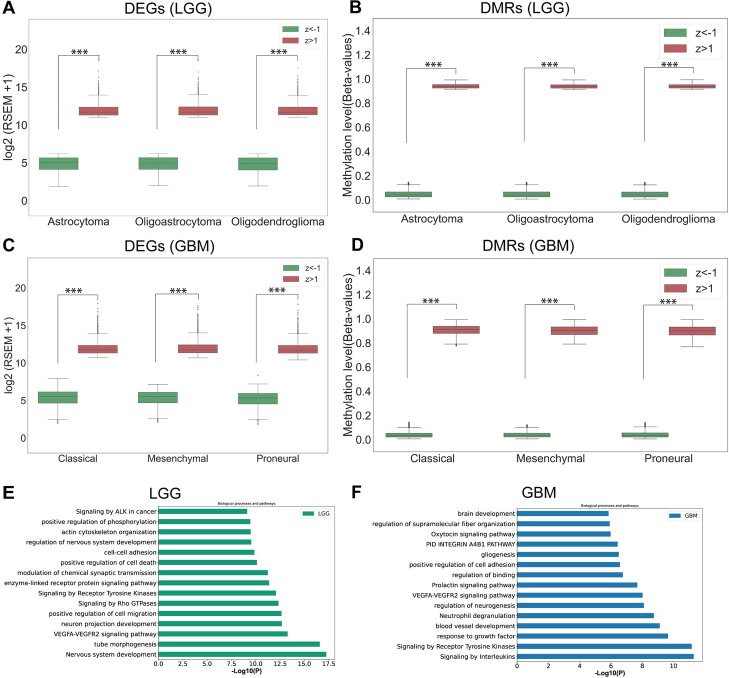
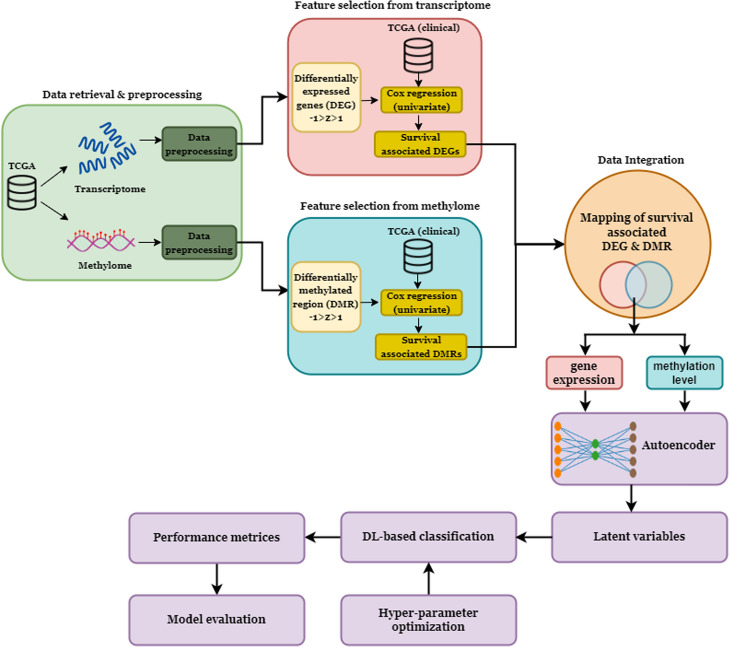
Results: Transcriptome and methylome data of glioma patients were preprocessed, and differentially expressed features from both datasets were identified. Subsequently, a Cox regression analysis determined genes and CpGs associated with survival. Gene set enrichment analysis was carried out to examine the biological significance of the features. Further, we identified CpG and gene pairs by mapping them in the promoter region of corresponding genes. The methylation and gene expression levels of these CpGs and genes were embedded in a lower-dimensional space with an autoencoder. Next, ANN and CNN were used to classify subtypes using the latent features from embedding space. CNN performs better than ANN for subtyping lower-grade gliomas (LGG) and glioblastoma multiforme (GBM). The subtyping accuracy of CNN was 98.03% (± 0.06) and 94.07% (± 0.01) in LGG and GBM, respectively. The precision of the models was 97.67% in LGG and 90.40% in GBM. The model sensitivity was 96.96% in LGG and 91.18% in GBM. Additionally, we observed the superior performance of CNN with external datasets. The genes and CpGs pairs used to develop the model showed better performance than the random CpGs-gene pairs, preprocessed data, and single omics data.
Conclusions: The current study showed that a novel feature selection and data integration strategy led to the development of DeepAutoGlioma, an effective framework for diagnosing glioma subtypes.
期刊介绍:
BioData Mining is an open access, open peer-reviewed journal encompassing research on all aspects of data mining applied to high-dimensional biological and biomedical data, focusing on computational aspects of knowledge discovery from large-scale genetic, transcriptomic, genomic, proteomic, and metabolomic data.
Topical areas include, but are not limited to:
-Development, evaluation, and application of novel data mining and machine learning algorithms.
-Adaptation, evaluation, and application of traditional data mining and machine learning algorithms.
-Open-source software for the application of data mining and machine learning algorithms.
-Design, development and integration of databases, software and web services for the storage, management, retrieval, and analysis of data from large scale studies.
-Pre-processing, post-processing, modeling, and interpretation of data mining and machine learning results for biological interpretation and knowledge discovery.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
