{"title":"Using DeepSignalingFlow to mine signaling flows interpreting mechanism of synergy of cocktails.","authors":"Heming Zhang, Yixin Chen, Philip Payne, Fuhai Li","doi":"10.1038/s41540-024-00421-w","DOIUrl":null,"url":null,"abstract":"<p><p>Complex signaling pathways are believed to be responsible for drug resistance. Drug combinations perturbing multiple signaling targets have the potential to reduce drug resistance. The large-scale multi-omic datasets and experimental drug combination synergistic score data are valuable resources to study mechanisms of synergy (MoS) to guide the development of precision drug combinations. However, signaling patterns of MoS are complex and remain unclear, and thus it is challenging to identify synergistic drug combinations in clinical. Herein, we proposed a novel integrative and interpretable graph AI model, DeepSignalingFlow, to uncover the MoS by integrating and mining multi-omic data. The major innovation is that we uncover MoS by modeling the signaling flow from multi-omic features of essential disease proteins to the drug targets, which has not been introduced by the existing models. The model performance was assessed utilizing four distinct drug combination synergy evaluation datasets, i.e., NCI ALMANAC, O'Neil, DrugComb, and DrugCombDB. The comparison results showed that the proposed model outperformed existing graph AI models in terms of synergy score prediction, and can interpret MoS using the core signaling flows. The code is publicly accessible via Github: https://github.com/FuhaiLiAiLab/DeepSignalingFlow.</p>","PeriodicalId":19345,"journal":{"name":"NPJ Systems Biology and Applications","volume":"10 1","pages":"92"},"PeriodicalIF":3.5000,"publicationDate":"2024-08-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11339460/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Systems Biology and Applications","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1038/s41540-024-00421-w","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
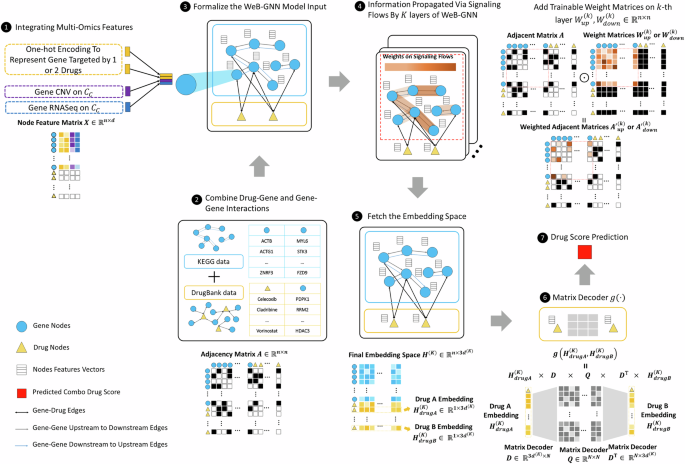
Complex signaling pathways are believed to be responsible for drug resistance. Drug combinations perturbing multiple signaling targets have the potential to reduce drug resistance. The large-scale multi-omic datasets and experimental drug combination synergistic score data are valuable resources to study mechanisms of synergy (MoS) to guide the development of precision drug combinations. However, signaling patterns of MoS are complex and remain unclear, and thus it is challenging to identify synergistic drug combinations in clinical. Herein, we proposed a novel integrative and interpretable graph AI model, DeepSignalingFlow, to uncover the MoS by integrating and mining multi-omic data. The major innovation is that we uncover MoS by modeling the signaling flow from multi-omic features of essential disease proteins to the drug targets, which has not been introduced by the existing models. The model performance was assessed utilizing four distinct drug combination synergy evaluation datasets, i.e., NCI ALMANAC, O'Neil, DrugComb, and DrugCombDB. The comparison results showed that the proposed model outperformed existing graph AI models in terms of synergy score prediction, and can interpret MoS using the core signaling flows. The code is publicly accessible via Github: https://github.com/FuhaiLiAiLab/DeepSignalingFlow.
期刊介绍:
npj Systems Biology and Applications is an online Open Access journal dedicated to publishing the premier research that takes a systems-oriented approach. The journal aims to provide a forum for the presentation of articles that help define this nascent field, as well as those that apply the advances to wider fields. We encourage studies that integrate, or aid the integration of, data, analyses and insight from molecules to organisms and broader systems. Important areas of interest include not only fundamental biological systems and drug discovery, but also applications to health, medical practice and implementation, big data, biotechnology, food science, human behaviour, broader biological systems and industrial applications of systems biology.
We encourage all approaches, including network biology, application of control theory to biological systems, computational modelling and analysis, comprehensive and/or high-content measurements, theoretical, analytical and computational studies of system-level properties of biological systems and computational/software/data platforms enabling such studies.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
