Emiliano Ventura-Macias, P. M. Martinez, Rubén Pérez, J. G. Vilhena
{"title":"Quantum Mechanical Derived (VdW-DFT) Transferable Lennard–Jones and Morse Potentials to Model Cysteine and Alkanethiol Adsorption on Au(111)","authors":"Emiliano Ventura-Macias, P. M. Martinez, Rubén Pérez, J. G. Vilhena","doi":"10.1002/admi.202400369","DOIUrl":null,"url":null,"abstract":"<p>The cysteine and alkanethiol adsorption on Au(111) surfaces is investigated using density functional theory (DFT) and classic molecular dynamics (MD). Understanding the S–Au interaction across different scales poses major challenges. DFT provides atomic-level precision but it hardly provides insight on nanosecond scale dynamics of this interface. Alternatively, MD, although it enables modeling larger systems for longer periods, its accuracy heavily relies on the parameterization of the force fields (FF). To address this, an MD potential is fitted using DFT calculations, bridging the gap in accuracy and efficiency. At the DFT level, it is found that PBE with DFT-D3 reproduces complex approaches at a fraction of the computational cost. Separating PBE and DFT-D3 contributions reveals consistent PBE energy across molecules (chemisorption), while dispersion varies (physisorption). Thus, the interaction energy of cysteine and two short-chain alkanethiols is calculated to parameterize both Morse and Lennard–Jones (LJ) potentials. The parameterization improves the potential energy in the preferred adsorption sites: the threefold hcp and fcc with respect to the previous proposals in the literature. Furthermore, the transferability is here demonstrated. At last, these results show that LJ potentials outperform more complex Morse potentials. The procedure is general, and the codes and supporting inputs are publicly available, allowing swift generation of potential energy surfaces (PES) at the DFT level, and fitted LJ or Morse potentials to any molecular interface.</p>","PeriodicalId":115,"journal":{"name":"Advanced Materials Interfaces","volume":"11 30","pages":""},"PeriodicalIF":4.4000,"publicationDate":"2024-08-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/admi.202400369","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advanced Materials Interfaces","FirstCategoryId":"88","ListUrlMain":"https://advanced.onlinelibrary.wiley.com/doi/10.1002/admi.202400369","RegionNum":3,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
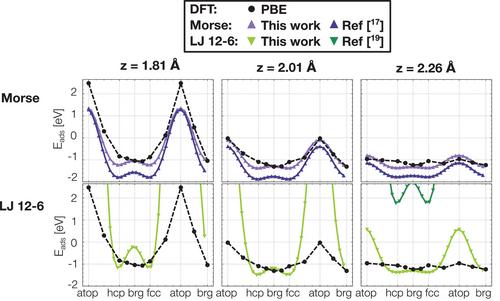
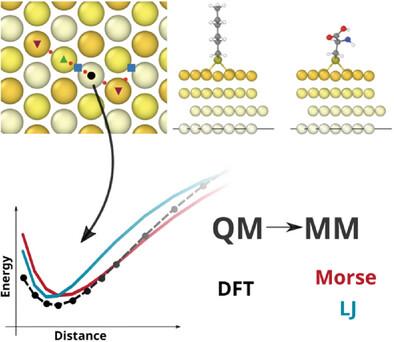
The cysteine and alkanethiol adsorption on Au(111) surfaces is investigated using density functional theory (DFT) and classic molecular dynamics (MD). Understanding the S–Au interaction across different scales poses major challenges. DFT provides atomic-level precision but it hardly provides insight on nanosecond scale dynamics of this interface. Alternatively, MD, although it enables modeling larger systems for longer periods, its accuracy heavily relies on the parameterization of the force fields (FF). To address this, an MD potential is fitted using DFT calculations, bridging the gap in accuracy and efficiency. At the DFT level, it is found that PBE with DFT-D3 reproduces complex approaches at a fraction of the computational cost. Separating PBE and DFT-D3 contributions reveals consistent PBE energy across molecules (chemisorption), while dispersion varies (physisorption). Thus, the interaction energy of cysteine and two short-chain alkanethiols is calculated to parameterize both Morse and Lennard–Jones (LJ) potentials. The parameterization improves the potential energy in the preferred adsorption sites: the threefold hcp and fcc with respect to the previous proposals in the literature. Furthermore, the transferability is here demonstrated. At last, these results show that LJ potentials outperform more complex Morse potentials. The procedure is general, and the codes and supporting inputs are publicly available, allowing swift generation of potential energy surfaces (PES) at the DFT level, and fitted LJ or Morse potentials to any molecular interface.
期刊介绍:
Advanced Materials Interfaces publishes top-level research on interface technologies and effects. Considering any interface formed between solids, liquids, and gases, the journal ensures an interdisciplinary blend of physics, chemistry, materials science, and life sciences. Advanced Materials Interfaces was launched in 2014 and received an Impact Factor of 4.834 in 2018.
The scope of Advanced Materials Interfaces is dedicated to interfaces and surfaces that play an essential role in virtually all materials and devices. Physics, chemistry, materials science and life sciences blend to encourage new, cross-pollinating ideas, which will drive forward our understanding of the processes at the interface.
Advanced Materials Interfaces covers all topics in interface-related research:
Oil / water separation,
Applications of nanostructured materials,
2D materials and heterostructures,
Surfaces and interfaces in organic electronic devices,
Catalysis and membranes,
Self-assembly and nanopatterned surfaces,
Composite and coating materials,
Biointerfaces for technical and medical applications.
Advanced Materials Interfaces provides a forum for topics on surface and interface science with a wide choice of formats: Reviews, Full Papers, and Communications, as well as Progress Reports and Research News.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
