Daniel F Thomas du Toit, Yuxing Zhou, Volker L Deringer
{"title":"Hyperparameter Optimization for Atomic Cluster Expansion Potentials.","authors":"Daniel F Thomas du Toit, Yuxing Zhou, Volker L Deringer","doi":"10.1021/acs.jctc.4c01012","DOIUrl":null,"url":null,"abstract":"<p><p>Machine learning-based interatomic potentials enable accurate materials simulations on extended time- and length scales. ML potentials based on the atomic cluster expansion (ACE) framework have recently shown promising performance for this purpose. Here, we describe a largely automated computational approach to optimizing hyperparameters for ACE potential models. We extend our openly available Python package, XPOT, to include an interface for ACE fitting, and discuss the optimization of the functional form and complexity of these models based on systematic sweeps across relevant hyperparameters. We showcase the usefulness of the approach for two example systems: the covalent network of silicon and the phase-change material Sb<sub>2</sub>Te<sub>3</sub>. More generally, our work emphasizes the importance of hyperparameter selection in the development of advanced ML potential models.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"10103-10113"},"PeriodicalIF":5.5000,"publicationDate":"2024-11-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11603601/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c01012","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/6 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
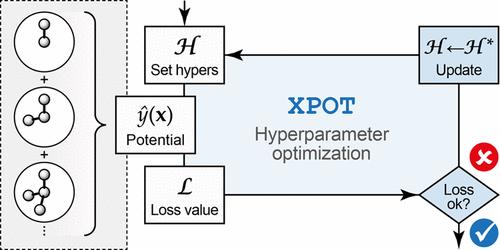
Machine learning-based interatomic potentials enable accurate materials simulations on extended time- and length scales. ML potentials based on the atomic cluster expansion (ACE) framework have recently shown promising performance for this purpose. Here, we describe a largely automated computational approach to optimizing hyperparameters for ACE potential models. We extend our openly available Python package, XPOT, to include an interface for ACE fitting, and discuss the optimization of the functional form and complexity of these models based on systematic sweeps across relevant hyperparameters. We showcase the usefulness of the approach for two example systems: the covalent network of silicon and the phase-change material Sb2Te3. More generally, our work emphasizes the importance of hyperparameter selection in the development of advanced ML potential models.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
