{"title":"Data efficient machine learning potentials for modeling catalytic reactivity via active learning and enhanced sampling","authors":"Simone Perego, Luigi Bonati","doi":"10.1038/s41524-024-01481-6","DOIUrl":null,"url":null,"abstract":"<p>Simulating catalytic reactivity under operative conditions poses a significant challenge due to the dynamic nature of the catalysts and the high computational cost of electronic structure calculations. Machine learning potentials offer a promising avenue to simulate dynamics at a fraction of the cost, but they require datasets containing all relevant configurations, particularly reactive ones. Here, we present a scheme to construct reactive potentials in a data-efficient manner. This is achieved by combining enhanced sampling methods first with Gaussian processes to discover transition paths and then with graph neural networks to obtain a uniformly accurate description. The necessary configurations are extracted via a Data-Efficient Active Learning (DEAL) procedure based on local environment uncertainty. We validated our approach by studying several reactions related to the decomposition of ammonia on iron-cobalt alloy catalysts. Our scheme proved to be efficient, requiring only ~1000 DFT calculations per reaction, and robust, sampling reactive configurations from the different accessible pathways. Using this potential, we calculated free energy profiles and characterized reaction mechanisms, showing the ability to provide microscopic insights into complex processes under dynamic conditions.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"23 1","pages":""},"PeriodicalIF":11.9000,"publicationDate":"2024-12-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01481-6","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
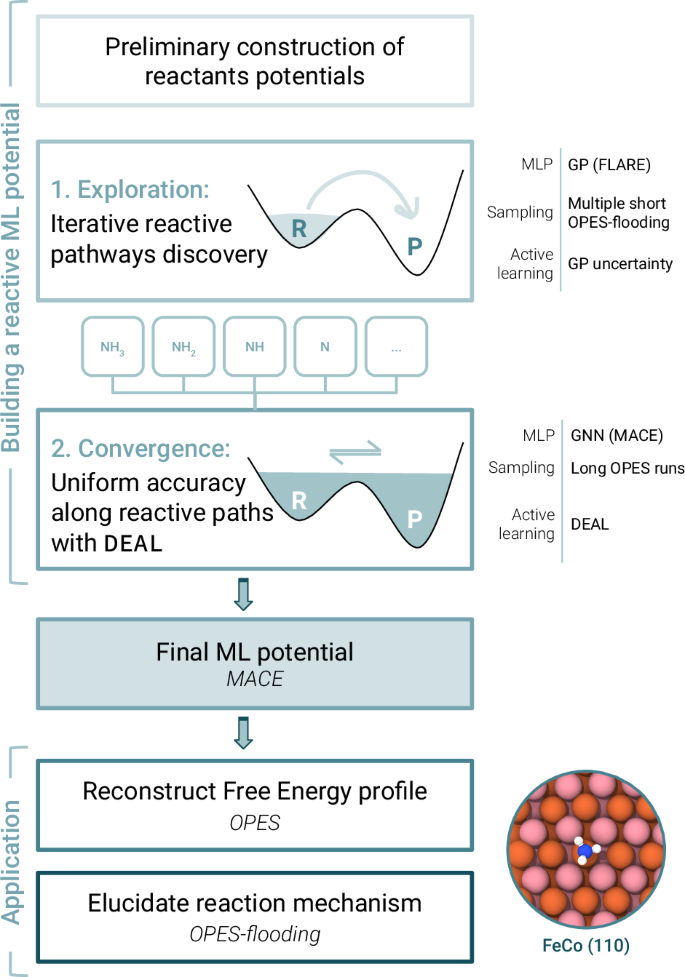
Abstract
Simulating catalytic reactivity under operative conditions poses a significant challenge due to the dynamic nature of the catalysts and the high computational cost of electronic structure calculations. Machine learning potentials offer a promising avenue to simulate dynamics at a fraction of the cost, but they require datasets containing all relevant configurations, particularly reactive ones. Here, we present a scheme to construct reactive potentials in a data-efficient manner. This is achieved by combining enhanced sampling methods first with Gaussian processes to discover transition paths and then with graph neural networks to obtain a uniformly accurate description. The necessary configurations are extracted via a Data-Efficient Active Learning (DEAL) procedure based on local environment uncertainty. We validated our approach by studying several reactions related to the decomposition of ammonia on iron-cobalt alloy catalysts. Our scheme proved to be efficient, requiring only ~1000 DFT calculations per reaction, and robust, sampling reactive configurations from the different accessible pathways. Using this potential, we calculated free energy profiles and characterized reaction mechanisms, showing the ability to provide microscopic insights into complex processes under dynamic conditions.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
