Aditya Venkatraman, Mark A. Wilson, David Montes de Oca Zapiain
{"title":"Accelerating charge estimation in molecular dynamics simulations using physics-informed neural networks: corrosion applications","authors":"Aditya Venkatraman, Mark A. Wilson, David Montes de Oca Zapiain","doi":"10.1038/s41524-024-01495-0","DOIUrl":null,"url":null,"abstract":"<p>Molecular Dynamics (MD) simulations are used to understand the effects of corrosion on metallic materials in salt brine. Reactive force fields in classical MD enable accurate modeling of bond formation and breakage in the aqueous medium and at the metal-electrolyte interface, while also facilitating dynamic partial charge equilibration. However, MD simulations are computationally intensive and unsuitable for modeling the long time scales characteristic of corrosive phenomena. To address this, we develop reduced-order machine learning models that provide accurate and efficient predictions of charge density in corrosive environments. Specifically, we use Long Short-Term Memory (LSTM) networks to forecast charge density evolution based on atomic environments represented by Smooth Overlap of Atomic Positions (SOAP) descriptors. A physics-informed loss function enforces charge neutrality and electronegativity equivalence. The atomic charges predicted by the deep learning model trained on this work were obtained two orders of magnitude faster than those from molecular dynamics (MD) simulations, with an error of less than 3% compared to the MD-obtained charges, even in extrapolative scenarios, while adhering to physical constraints. This demonstrates the excellent accuracy, computational efficiency, and validity of the developed model. Lastly, even though developed for corrosion, these protocols are formulated in a phenomenon-agnostic manner, allowing application to various variable-charge interatomic potentials and related fields.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"77 2 1","pages":""},"PeriodicalIF":11.9000,"publicationDate":"2025-02-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01495-0","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
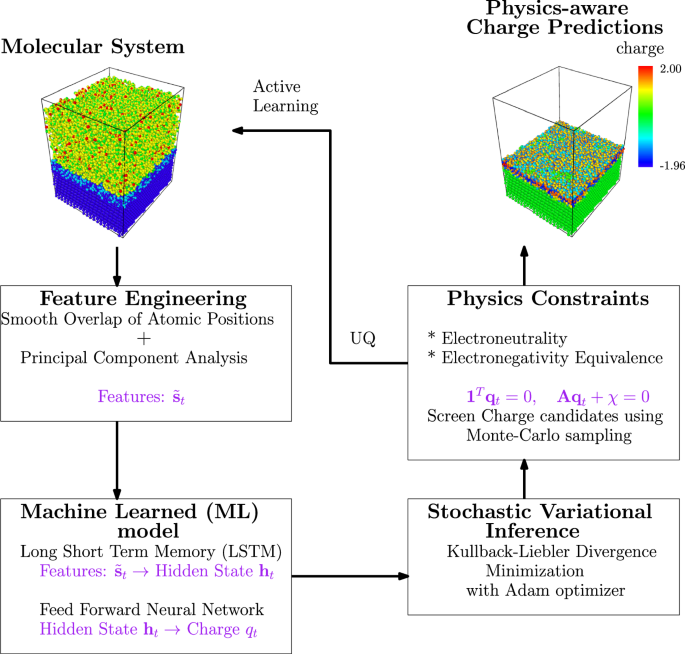
Molecular Dynamics (MD) simulations are used to understand the effects of corrosion on metallic materials in salt brine. Reactive force fields in classical MD enable accurate modeling of bond formation and breakage in the aqueous medium and at the metal-electrolyte interface, while also facilitating dynamic partial charge equilibration. However, MD simulations are computationally intensive and unsuitable for modeling the long time scales characteristic of corrosive phenomena. To address this, we develop reduced-order machine learning models that provide accurate and efficient predictions of charge density in corrosive environments. Specifically, we use Long Short-Term Memory (LSTM) networks to forecast charge density evolution based on atomic environments represented by Smooth Overlap of Atomic Positions (SOAP) descriptors. A physics-informed loss function enforces charge neutrality and electronegativity equivalence. The atomic charges predicted by the deep learning model trained on this work were obtained two orders of magnitude faster than those from molecular dynamics (MD) simulations, with an error of less than 3% compared to the MD-obtained charges, even in extrapolative scenarios, while adhering to physical constraints. This demonstrates the excellent accuracy, computational efficiency, and validity of the developed model. Lastly, even though developed for corrosion, these protocols are formulated in a phenomenon-agnostic manner, allowing application to various variable-charge interatomic potentials and related fields.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
