{"title":"CTNNB1 syndrome mouse models.","authors":"Duško Lainšček, Vida Forstnerič, Špela Miroševič","doi":"10.1007/s00335-025-10105-3","DOIUrl":null,"url":null,"abstract":"<p><p>CTNNB1 syndrome is a rare neurodevelopmental disorder, affecting children worldwide with a prevalence of 2.6-3.2 per 100,000 births and often misdiagnosed as cerebral palsy. De novo loss-of-function mutations in the Ctnnb1 gene result in dysfunction of the β-catenin protein, disrupting the canonical Wnt signaling pathway, which plays a key role in cell proliferation, differentiation, and tissue homeostasis. Additionally, these mutations impair the formation of cell junctions, adversely affecting tissue architecture. Motor and speech deficits, cognitive impairment, cardiovascular and visual problems are just some of the key symptoms that occur in CTNNB1 syndrome patients. There is currently no effective treatment option available for patients with CTNNB1 syndrome, with support largely focused on the management of symptoms and physiotherapy, yet recently some therapeutic approaches are being developed. Animal testing is still crucial in the process of new drug development, and mouse models are particularly important. These models provide researchers with new understanding of the disease mechanisms and are invaluable for testing the efficacy and safety of potential treatments. The development of various mouse models with β-catenin loss- and gain-of-function mutations successfully replicates key features of intellectual disability, autism-like behaviors, motor deficits, and more. These models provide a valuable platform for studying disease mechanisms and offer a powerful tool for testing the therapeutic potential and effectiveness of new drug candidates, paving the way for future clinical trials.</p>","PeriodicalId":18259,"journal":{"name":"Mammalian Genome","volume":" ","pages":"390-402"},"PeriodicalIF":2.7000,"publicationDate":"2025-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12130087/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Mammalian Genome","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00335-025-10105-3","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/20 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
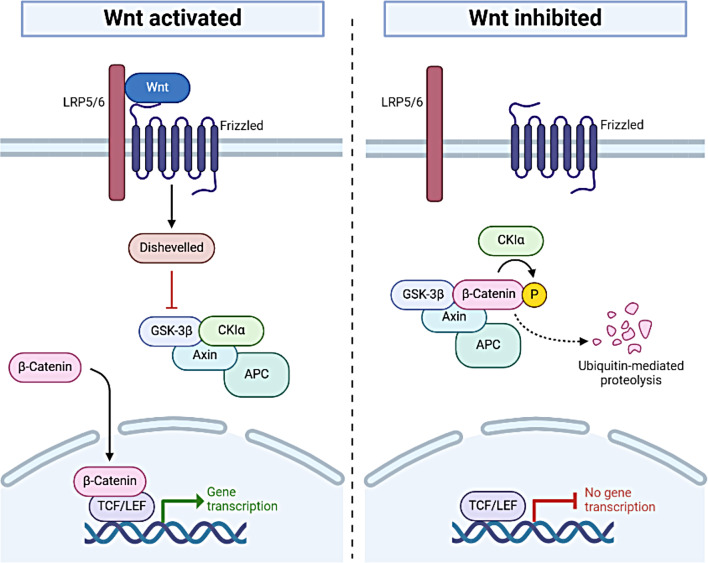
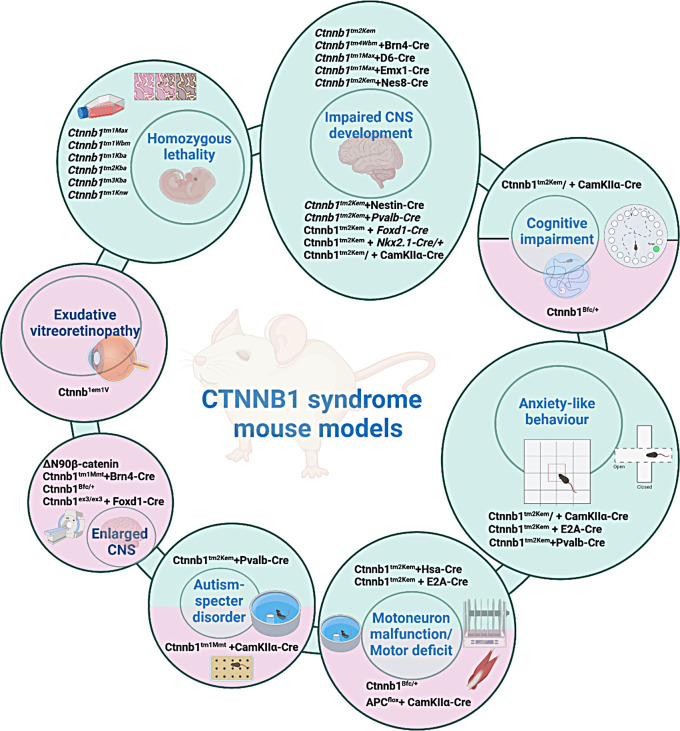
CTNNB1 syndrome is a rare neurodevelopmental disorder, affecting children worldwide with a prevalence of 2.6-3.2 per 100,000 births and often misdiagnosed as cerebral palsy. De novo loss-of-function mutations in the Ctnnb1 gene result in dysfunction of the β-catenin protein, disrupting the canonical Wnt signaling pathway, which plays a key role in cell proliferation, differentiation, and tissue homeostasis. Additionally, these mutations impair the formation of cell junctions, adversely affecting tissue architecture. Motor and speech deficits, cognitive impairment, cardiovascular and visual problems are just some of the key symptoms that occur in CTNNB1 syndrome patients. There is currently no effective treatment option available for patients with CTNNB1 syndrome, with support largely focused on the management of symptoms and physiotherapy, yet recently some therapeutic approaches are being developed. Animal testing is still crucial in the process of new drug development, and mouse models are particularly important. These models provide researchers with new understanding of the disease mechanisms and are invaluable for testing the efficacy and safety of potential treatments. The development of various mouse models with β-catenin loss- and gain-of-function mutations successfully replicates key features of intellectual disability, autism-like behaviors, motor deficits, and more. These models provide a valuable platform for studying disease mechanisms and offer a powerful tool for testing the therapeutic potential and effectiveness of new drug candidates, paving the way for future clinical trials.
期刊介绍:
Mammalian Genome focuses on the experimental, theoretical and technical aspects of genetics, genomics, epigenetics and systems biology in mouse, human and other mammalian species, with an emphasis on the relationship between genotype and phenotype, elucidation of biological and disease pathways as well as experimental aspects of interventions, therapeutics, and precision medicine. The journal aims to publish high quality original papers that present novel findings in all areas of mammalian genetic research as well as review articles on areas of topical interest. The journal will also feature commentaries and editorials to inform readers of breakthrough discoveries as well as issues of research standards, policies and ethics.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
