{"title":"Dynamic integration of feature- and template-based methods improves the prediction of conformational B cell epitopes","authors":"Yueyue Shen, Zheng Jiang, Rong Liu","doi":"10.1016/j.str.2025.01.018","DOIUrl":null,"url":null,"abstract":"The accurate prediction of conformational epitopes promotes our understanding of antigen-antibody interactions. All existing algorithms depend on a feature-based strategy, which limits their performance. A template-based strategy can provide complementary information, and the interplay between these two strategies could improve the prediction of epitopes. Here, we present DynaBCE, a dynamic ensemble algorithm to effectively identify conformational B cell epitopes (BCEs). Using novel handcrafted structural descriptors and embeddings from protein language models, we developed machine learning and deep learning modules based on boosting algorithms and geometric graph neural networks, respectively. Furthermore, we built a template module by leveraging known structural template information and transformer-based algorithms to capture binding signatures. Finally, we integrated the three modules using a dynamic weighting approach to maximize the strength of each module for different samples. DynaBCE achieved promising results for both native and predicted structures and outperformed previous methods as demonstrated in various evaluation scenarios.","PeriodicalId":22168,"journal":{"name":"Structure","volume":"28 1","pages":""},"PeriodicalIF":4.3000,"publicationDate":"2025-02-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Structure","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1016/j.str.2025.01.018","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
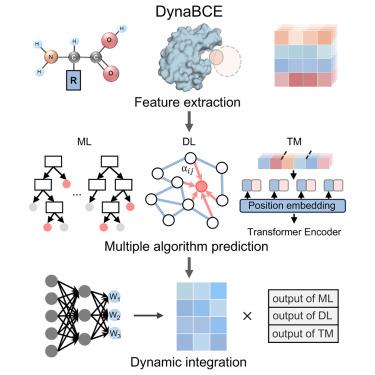
The accurate prediction of conformational epitopes promotes our understanding of antigen-antibody interactions. All existing algorithms depend on a feature-based strategy, which limits their performance. A template-based strategy can provide complementary information, and the interplay between these two strategies could improve the prediction of epitopes. Here, we present DynaBCE, a dynamic ensemble algorithm to effectively identify conformational B cell epitopes (BCEs). Using novel handcrafted structural descriptors and embeddings from protein language models, we developed machine learning and deep learning modules based on boosting algorithms and geometric graph neural networks, respectively. Furthermore, we built a template module by leveraging known structural template information and transformer-based algorithms to capture binding signatures. Finally, we integrated the three modules using a dynamic weighting approach to maximize the strength of each module for different samples. DynaBCE achieved promising results for both native and predicted structures and outperformed previous methods as demonstrated in various evaluation scenarios.
期刊介绍:
Structure aims to publish papers of exceptional interest in the field of structural biology. The journal strives to be essential reading for structural biologists, as well as biologists and biochemists that are interested in macromolecular structure and function. Structure strongly encourages the submission of manuscripts that present structural and molecular insights into biological function and mechanism. Other reports that address fundamental questions in structural biology, such as structure-based examinations of protein evolution, folding, and/or design, will also be considered. We will consider the application of any method, experimental or computational, at high or low resolution, to conduct structural investigations, as long as the method is appropriate for the biological, functional, and mechanistic question(s) being addressed. Likewise, reports describing single-molecule analysis of biological mechanisms are welcome.
In general, the editors encourage submission of experimental structural studies that are enriched by an analysis of structure-activity relationships and will not consider studies that solely report structural information unless the structure or analysis is of exceptional and broad interest. Studies reporting only homology models, de novo models, or molecular dynamics simulations are also discouraged unless the models are informed by or validated by novel experimental data; rationalization of a large body of existing experimental evidence and making testable predictions based on a model or simulation is often not considered sufficient.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
