{"title":"Sweetener aspartame aggravates atherosclerosis through insulin-triggered inflammation","authors":"Weijie Wu, Wenhai Sui, Sizhe Chen, Ziheng Guo, Xu Jing, Xiaolu Wang, Qun Wang, Xinshuang Yu, Wenjing Xiong, Jiansong Ji, Libo Yang, Yuan Zhang, Wenjing Jiang, Guohua Yu, Shuzhen Liu, Wei Tao, Chen Zhao, Yun Zhang, Yuguo Chen, Cheng Zhang, Yihai Cao","doi":"10.1016/j.cmet.2025.01.006","DOIUrl":null,"url":null,"abstract":"Consumption of artificial sweeteners (ASWs) in various foods and beverages has been linked to an increased risk of cardiovascular diseases (CVDs). However, molecular mechanisms underlying ASW-associated CVD remain unknown. Here, we show that consumption of 0.15% aspartame (APM) markedly increased insulin secretion in mice and monkeys. Bilateral subdiaphragmatic vagotomy (SDV) obliterated APM-elevated blood insulin levels, demonstrating crucial roles of parasympathetic activation in regulation of insulin secretion. Incessant APM feeding of ApoE<sup>−/</sup><sup>−</sup> mice aggravated atherosclerotic plaque formation and growth via an insulin-dependent mechanism. Implantation of an insulin-slow-release pump in ApoE<sup>−/−</sup> mice exacerbated atherosclerosis. Whole-genome expression profiling discovered that CX3CL1 chemokine was the most upregulated gene in the insulin-stimulated arterial endothelial cells. Specific deletion of a CX3CL1 receptor, <em>Cx3cr1</em> gene, in monocytes/macrophages completely abrogated the APM-exacerbated atherosclerosis. Our findings uncover a novel mechanism of APM-associated atherosclerosis and therapeutic targeting of the endothelial CX3CL1-macrophage CX3CR1 signaling axis provides an approach for treating atherosclerotic CVD.","PeriodicalId":9840,"journal":{"name":"Cell metabolism","volume":"2 1","pages":""},"PeriodicalIF":30.9000,"publicationDate":"2025-02-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell metabolism","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1016/j.cmet.2025.01.006","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
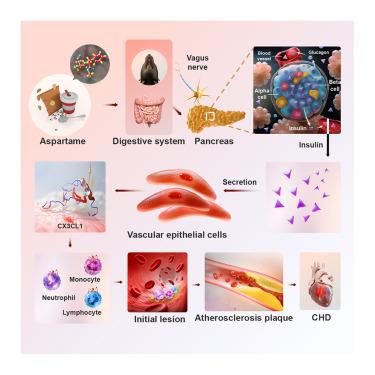
Consumption of artificial sweeteners (ASWs) in various foods and beverages has been linked to an increased risk of cardiovascular diseases (CVDs). However, molecular mechanisms underlying ASW-associated CVD remain unknown. Here, we show that consumption of 0.15% aspartame (APM) markedly increased insulin secretion in mice and monkeys. Bilateral subdiaphragmatic vagotomy (SDV) obliterated APM-elevated blood insulin levels, demonstrating crucial roles of parasympathetic activation in regulation of insulin secretion. Incessant APM feeding of ApoE−/− mice aggravated atherosclerotic plaque formation and growth via an insulin-dependent mechanism. Implantation of an insulin-slow-release pump in ApoE−/− mice exacerbated atherosclerosis. Whole-genome expression profiling discovered that CX3CL1 chemokine was the most upregulated gene in the insulin-stimulated arterial endothelial cells. Specific deletion of a CX3CL1 receptor, Cx3cr1 gene, in monocytes/macrophages completely abrogated the APM-exacerbated atherosclerosis. Our findings uncover a novel mechanism of APM-associated atherosclerosis and therapeutic targeting of the endothelial CX3CL1-macrophage CX3CR1 signaling axis provides an approach for treating atherosclerotic CVD.
期刊介绍:
Cell Metabolism is a top research journal established in 2005 that focuses on publishing original and impactful papers in the field of metabolic research.It covers a wide range of topics including diabetes, obesity, cardiovascular biology, aging and stress responses, circadian biology, and many others.
Cell Metabolism aims to contribute to the advancement of metabolic research by providing a platform for the publication and dissemination of high-quality research and thought-provoking articles.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
