Marco Barresi, Giulia Dal Santo, Rossella Izzo, Andrea Zauli, Eleonora Lamantea, Leonardo Caporali, Daniele Ghezzi, Andrea Legati
{"title":"Bioinformatics Tools for NGS-Based Identification of Single Nucleotide Variants and Large-Scale Rearrangements in Mitochondrial DNA.","authors":"Marco Barresi, Giulia Dal Santo, Rossella Izzo, Andrea Zauli, Eleonora Lamantea, Leonardo Caporali, Daniele Ghezzi, Andrea Legati","doi":"10.3390/biotech14010009","DOIUrl":null,"url":null,"abstract":"<p><p>The unique features of mitochondrial DNA (mtDNA), including its circular and multicopy nature, the possible coexistence of wild-type and mutant molecules (i.e., heteroplasmy) and the presence of nuclear mitochondrial DNA segments (NUMTs), make the diagnosis of mtDNA diseases particularly challenging. The extensive deployment of next-generation sequencing (NGS) technologies has significantly advanced the diagnosis of mtDNA-related diseases. However, the vast amounts and diverse types of sequencing data complicate the interpretation of these variants. From sequence alignment to variant calling, NGS-based mtDNA sequencing requires specialized bioinformatics tools, adapted for the mitochondrial genome. This study presents the use of new bioinformatics approaches, optimized for short- and long-read sequencing data, to enhance the accuracy of mtDNA analysis in diagnostics. Two recent and emerging free bioinformatics tools, Mitopore and MitoSAlt, were evaluated on patients previously diagnosed with single nucleotide variants or large-scale deletions. Analyses were performed in Linux-based environments and web servers implemented in Python, Perl, Java, and R. The results indicated that each tool demonstrated high sensitivity and specific accuracy in identifying and quantifying various types of pathogenic variants. The study suggests that the integrated and parallel use of these tools offers a significant advantage over traditional methods in interpreting mtDNA genetic variants, reducing the computational demands, and provides an accurate diagnostic solution.</p>","PeriodicalId":34490,"journal":{"name":"BioTech","volume":"14 1","pages":""},"PeriodicalIF":3.1000,"publicationDate":"2025-02-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11843820/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BioTech","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/biotech14010009","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
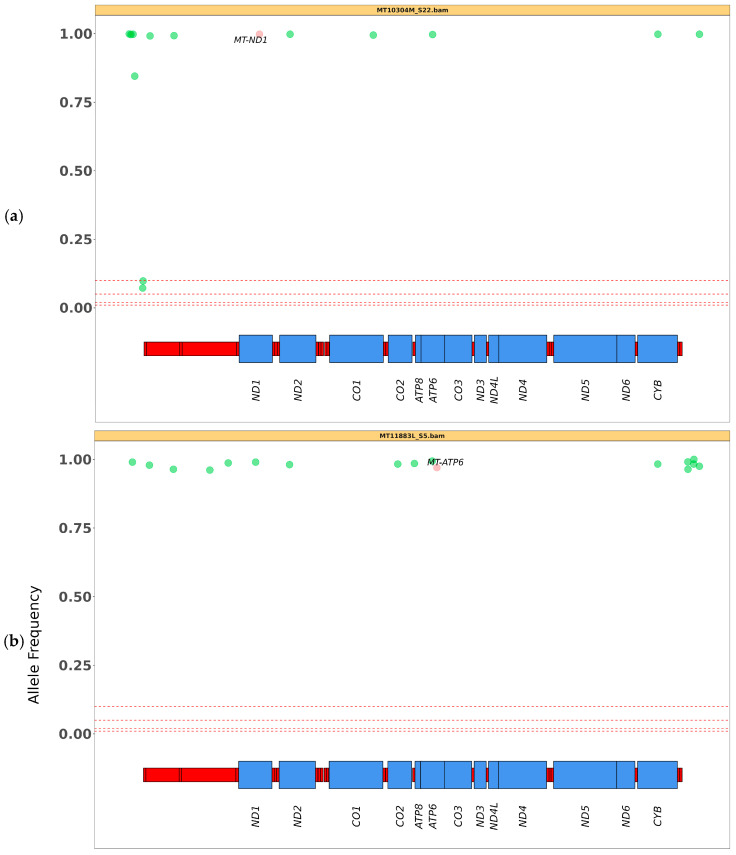
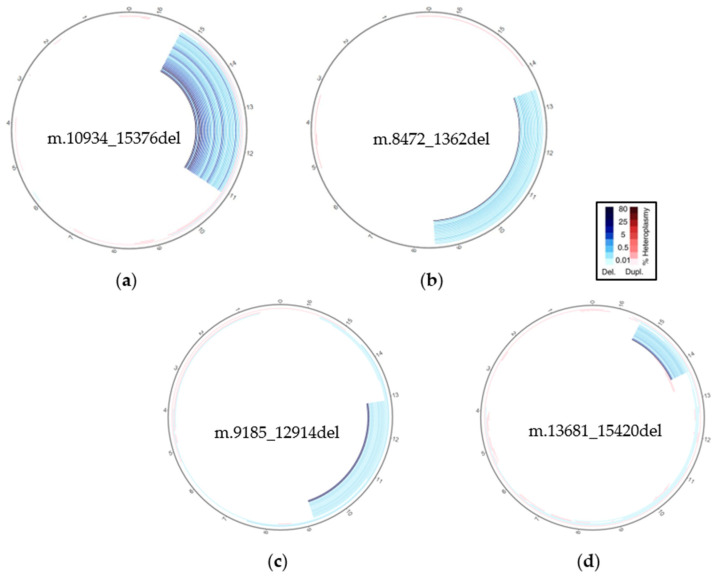
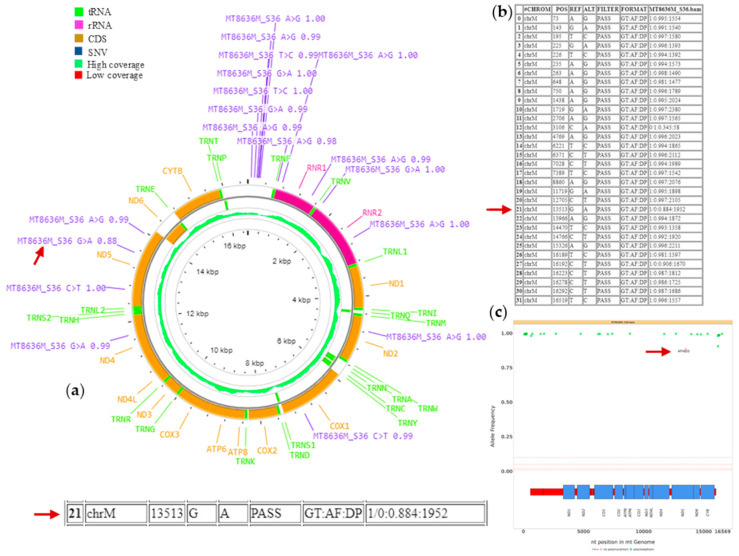
The unique features of mitochondrial DNA (mtDNA), including its circular and multicopy nature, the possible coexistence of wild-type and mutant molecules (i.e., heteroplasmy) and the presence of nuclear mitochondrial DNA segments (NUMTs), make the diagnosis of mtDNA diseases particularly challenging. The extensive deployment of next-generation sequencing (NGS) technologies has significantly advanced the diagnosis of mtDNA-related diseases. However, the vast amounts and diverse types of sequencing data complicate the interpretation of these variants. From sequence alignment to variant calling, NGS-based mtDNA sequencing requires specialized bioinformatics tools, adapted for the mitochondrial genome. This study presents the use of new bioinformatics approaches, optimized for short- and long-read sequencing data, to enhance the accuracy of mtDNA analysis in diagnostics. Two recent and emerging free bioinformatics tools, Mitopore and MitoSAlt, were evaluated on patients previously diagnosed with single nucleotide variants or large-scale deletions. Analyses were performed in Linux-based environments and web servers implemented in Python, Perl, Java, and R. The results indicated that each tool demonstrated high sensitivity and specific accuracy in identifying and quantifying various types of pathogenic variants. The study suggests that the integrated and parallel use of these tools offers a significant advantage over traditional methods in interpreting mtDNA genetic variants, reducing the computational demands, and provides an accurate diagnostic solution.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
