Irving Estevez, Benjamin D. Buckley, Marissa Lindman, Nicholas Panzera, Tsui-Wen Chou, Micheal McCourt, Brandon J. Vaglio, Colm Atkins, Bonnie L. Firestein, Brian P. Daniels
{"title":"The kinase RIPK3 promotes neuronal survival by suppressing excitatory neurotransmission during central nervous system viral infection","authors":"Irving Estevez, Benjamin D. Buckley, Marissa Lindman, Nicholas Panzera, Tsui-Wen Chou, Micheal McCourt, Brandon J. Vaglio, Colm Atkins, Bonnie L. Firestein, Brian P. Daniels","doi":"10.1016/j.immuni.2025.01.017","DOIUrl":null,"url":null,"abstract":"While recent work has identified roles for immune mediators in regulating neural activity, how innate immune signaling within neurons influences neurotransmission remains poorly understood. Emerging evidence suggests that the modulation of neurotransmission may serve important roles in host protection during infection of the central nervous system. Here, we showed that receptor-interacting protein kinase-3 (RIPK3) preserved neuronal survival during flavivirus infection through the suppression of excitatory neurotransmission. These effects occurred independently of the traditional functions of RIPK3 in promoting necroptosis and inflammatory transcription. Instead, RIPK3 promoted phosphorylation of the neuronal regulatory kinase calcium/calmodulin-dependent protein kinase II (CaMKII), which in turn activated the transcription factor cyclic AMP response element-binding protein (CREB) to drive a neuroprotective transcriptional program and suppress deleterious glutamatergic signaling. These findings identify an unexpected function for a canonical cell death protein in promoting neuronal survival during viral infection through the modulation of neuronal activity, highlighting mechanisms of neuroimmune crosstalk.","PeriodicalId":13269,"journal":{"name":"Immunity","volume":"30 1","pages":""},"PeriodicalIF":25.5000,"publicationDate":"2025-02-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Immunity","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1016/j.immuni.2025.01.017","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
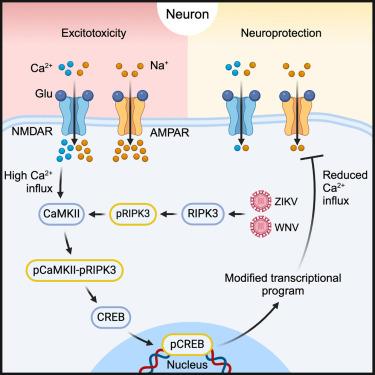
While recent work has identified roles for immune mediators in regulating neural activity, how innate immune signaling within neurons influences neurotransmission remains poorly understood. Emerging evidence suggests that the modulation of neurotransmission may serve important roles in host protection during infection of the central nervous system. Here, we showed that receptor-interacting protein kinase-3 (RIPK3) preserved neuronal survival during flavivirus infection through the suppression of excitatory neurotransmission. These effects occurred independently of the traditional functions of RIPK3 in promoting necroptosis and inflammatory transcription. Instead, RIPK3 promoted phosphorylation of the neuronal regulatory kinase calcium/calmodulin-dependent protein kinase II (CaMKII), which in turn activated the transcription factor cyclic AMP response element-binding protein (CREB) to drive a neuroprotective transcriptional program and suppress deleterious glutamatergic signaling. These findings identify an unexpected function for a canonical cell death protein in promoting neuronal survival during viral infection through the modulation of neuronal activity, highlighting mechanisms of neuroimmune crosstalk.
期刊介绍:
Immunity is a publication that focuses on publishing significant advancements in research related to immunology. We encourage the submission of studies that offer groundbreaking immunological discoveries, whether at the molecular, cellular, or whole organism level. Topics of interest encompass a wide range, such as cancer, infectious diseases, neuroimmunology, autoimmune diseases, allergies, mucosal immunity, metabolic diseases, and homeostasis.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
