Ruchir Rastogi, Ryan Chung, Sindy Li, Chang Li, Kyoungyeul Lee, Junwoo Woo, Dong-Wook Kim, Changwon Keum, Giulia Babbi, Pier Luigi Martelli, Castrense Savojardo, Rita Casadio, Kirsley Chennen, Thomas Weber, Olivier Poch, François Ancien, Gabriel Cia, Fabrizio Pucci, Daniele Raimondi, Wim Vranken, Marianne Rooman, Céline Marquet, Tobias Olenyi, Burkhard Rost, Gaia Andreoletti, Akash Kamandula, Yisu Peng, Constantina Bakolitsa, Matthew Mort, David N Cooper, Timothy Bergquist, Vikas Pejaver, Xiaoming Liu, Predrag Radivojac, Steven E Brenner, Nilah M Ioannidis
{"title":"Critical assessment of missense variant effect predictors on disease-relevant variant data.","authors":"Ruchir Rastogi, Ryan Chung, Sindy Li, Chang Li, Kyoungyeul Lee, Junwoo Woo, Dong-Wook Kim, Changwon Keum, Giulia Babbi, Pier Luigi Martelli, Castrense Savojardo, Rita Casadio, Kirsley Chennen, Thomas Weber, Olivier Poch, François Ancien, Gabriel Cia, Fabrizio Pucci, Daniele Raimondi, Wim Vranken, Marianne Rooman, Céline Marquet, Tobias Olenyi, Burkhard Rost, Gaia Andreoletti, Akash Kamandula, Yisu Peng, Constantina Bakolitsa, Matthew Mort, David N Cooper, Timothy Bergquist, Vikas Pejaver, Xiaoming Liu, Predrag Radivojac, Steven E Brenner, Nilah M Ioannidis","doi":"10.1007/s00439-025-02732-2","DOIUrl":null,"url":null,"abstract":"<p><p>Regular, systematic, and independent assessments of computational tools that are used to predict the pathogenicity of missense variants are necessary to evaluate their clinical and research utility and guide future improvements. The Critical Assessment of Genome Interpretation (CAGI) conducts the ongoing Annotate-All-Missense (Missense Marathon) challenge, in which missense variant effect predictors (also called variant impact predictors) are evaluated on missense variants added to disease-relevant databases following the prediction submission deadline. Here we assess predictors submitted to the CAGI 6 Annotate-All-Missense challenge, predictors commonly used in clinical genetics, and recently developed deep learning methods. We examine performance across a range of settings relevant for clinical and research applications, focusing on different subsets of the evaluation data as well as high-specificity and high-sensitivity regimes. Our evaluations reveal notable advances in current methods relative to older, well-cited tools in the field. While meta-predictors tend to outperform their constituent individual predictors, several newer individual predictors perform comparably to commonly used meta-predictors. Predictor performance varies between high-specificity and high-sensitivity regimes, highlighting that different methods may be optimal for different use cases. We also characterize two potential sources of bias. Predictors that incorporate allele frequency as a predictive feature tend to have reduced performance when distinguishing pathogenic variants from very rare benign variants, and predictors trained on pathogenicity labels from curated variant databases often inherit gene-level label imbalances. Our findings help illuminate the clinical and research utility of modern missense variant effect predictors and identify potential areas for future development.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":" ","pages":"281-293"},"PeriodicalIF":3.6000,"publicationDate":"2025-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11976771/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-025-02732-2","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/3/21 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
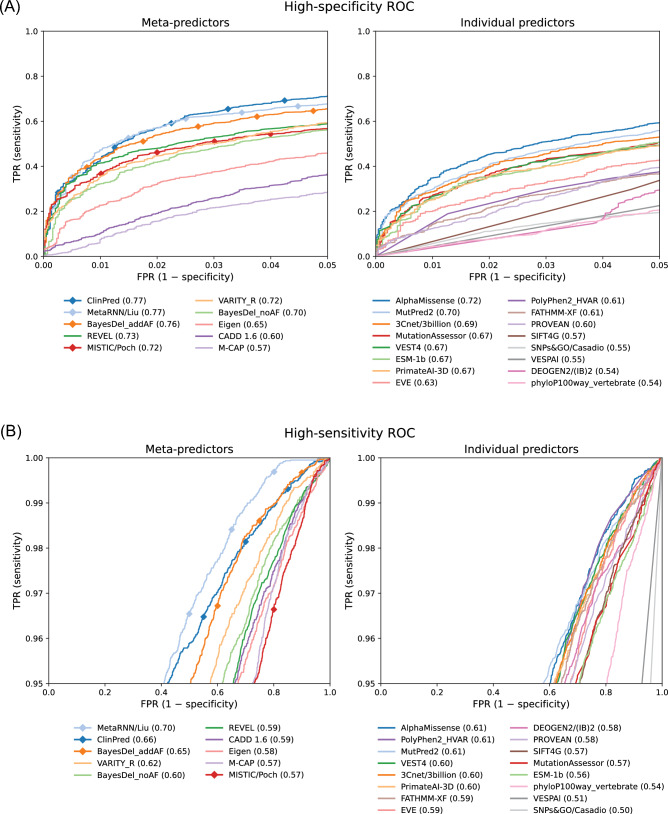
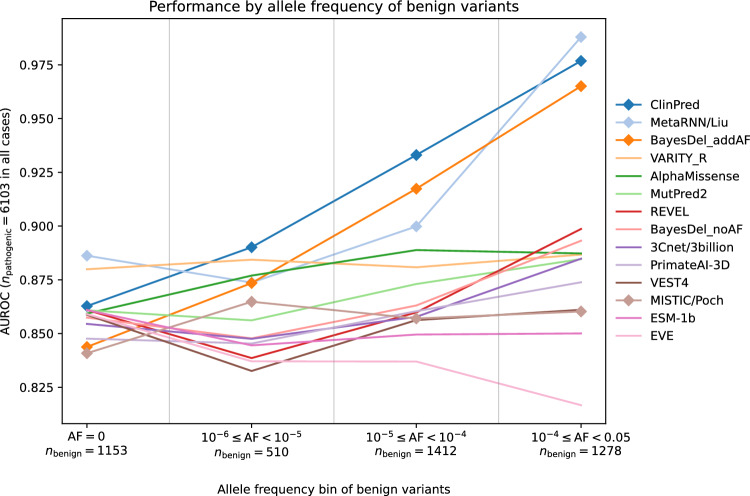
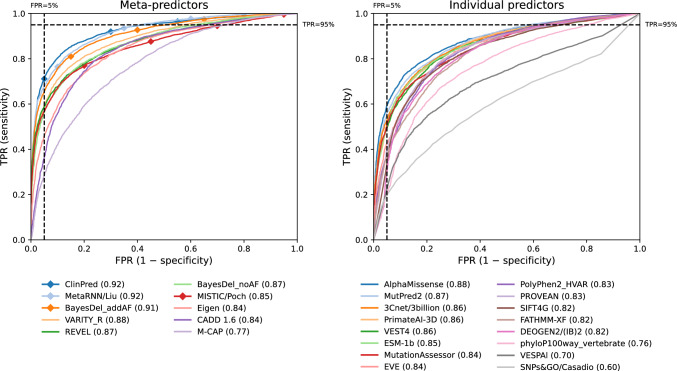
Regular, systematic, and independent assessments of computational tools that are used to predict the pathogenicity of missense variants are necessary to evaluate their clinical and research utility and guide future improvements. The Critical Assessment of Genome Interpretation (CAGI) conducts the ongoing Annotate-All-Missense (Missense Marathon) challenge, in which missense variant effect predictors (also called variant impact predictors) are evaluated on missense variants added to disease-relevant databases following the prediction submission deadline. Here we assess predictors submitted to the CAGI 6 Annotate-All-Missense challenge, predictors commonly used in clinical genetics, and recently developed deep learning methods. We examine performance across a range of settings relevant for clinical and research applications, focusing on different subsets of the evaluation data as well as high-specificity and high-sensitivity regimes. Our evaluations reveal notable advances in current methods relative to older, well-cited tools in the field. While meta-predictors tend to outperform their constituent individual predictors, several newer individual predictors perform comparably to commonly used meta-predictors. Predictor performance varies between high-specificity and high-sensitivity regimes, highlighting that different methods may be optimal for different use cases. We also characterize two potential sources of bias. Predictors that incorporate allele frequency as a predictive feature tend to have reduced performance when distinguishing pathogenic variants from very rare benign variants, and predictors trained on pathogenicity labels from curated variant databases often inherit gene-level label imbalances. Our findings help illuminate the clinical and research utility of modern missense variant effect predictors and identify potential areas for future development.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
