Xubo Hu, Kiet T. Nguyen, Vernon C. Jiang, Denene Lofland, Heinz E. Moser, Dehua Pei
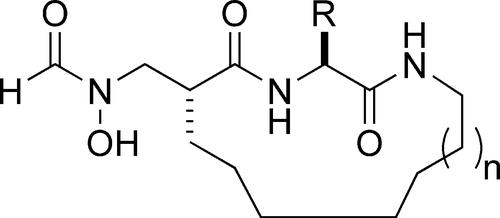
{"title":"Macrocyclic Inhibitors for Peptide Deformylase: A Structure−Activity Relationship Study of the Ring Size","authors":"Xubo Hu, Kiet T. Nguyen, Vernon C. Jiang, Denene Lofland, Heinz E. Moser, Dehua Pei","doi":"10.1021/jm049592c","DOIUrl":null,"url":null,"abstract":"<p >Peptide deformylase (PDF) catalyzes the removal of the N-terminal formyl group from newly synthesized polypeptides in eubacteria. Its essential role in bacterial cells but not in mammalian cells makes it an attractive target for antibacterial drug design. We have previously reported an N-formylhydroxylamine-based, metal-chelating macrocyclic PDF inhibitor, in which the P<sub>1</sub>‘ and P<sub>3</sub>‘ side chains are covalently joined. In this work, we have carried out a structure?activity relationship study on the size of the macrocycle and found that 15?17-membered macrocycles are optimal for binding to the PDF active site. Unlike the acyclic compounds, which are simple competitive inhibitors, the cyclic compounds all act as slow-binding inhibitors. As compared to their acyclic counterparts, the cyclic inhibitors displayed 20?50-fold higher potency against the PDF active site (<i>K</i><sub>I</sub>* as low as 70 pM), improved selectivity toward PDF, and improved the metabolic stability in rat plasma. Some of the macrocyclic inhibitors had potent, broad spectrum antibacterial activity against clinically significant Gram-positive and Gram-negative pathogens. These results suggest that the macrocyclic scaffold provides an excellent lead for the development of a new class of antibiotics. </p>","PeriodicalId":46,"journal":{"name":"Journal of Medicinal Chemistry","volume":"47 20","pages":"4941–4949"},"PeriodicalIF":6.8000,"publicationDate":"2004-08-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1021/jm049592c","citationCount":"41","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Medicinal Chemistry","FirstCategoryId":"3","ListUrlMain":"https://pubs.acs.org/doi/10.1021/jm049592c","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 41
Abstract
Peptide deformylase (PDF) catalyzes the removal of the N-terminal formyl group from newly synthesized polypeptides in eubacteria. Its essential role in bacterial cells but not in mammalian cells makes it an attractive target for antibacterial drug design. We have previously reported an N-formylhydroxylamine-based, metal-chelating macrocyclic PDF inhibitor, in which the P1‘ and P3‘ side chains are covalently joined. In this work, we have carried out a structure?activity relationship study on the size of the macrocycle and found that 15?17-membered macrocycles are optimal for binding to the PDF active site. Unlike the acyclic compounds, which are simple competitive inhibitors, the cyclic compounds all act as slow-binding inhibitors. As compared to their acyclic counterparts, the cyclic inhibitors displayed 20?50-fold higher potency against the PDF active site (KI* as low as 70 pM), improved selectivity toward PDF, and improved the metabolic stability in rat plasma. Some of the macrocyclic inhibitors had potent, broad spectrum antibacterial activity against clinically significant Gram-positive and Gram-negative pathogens. These results suggest that the macrocyclic scaffold provides an excellent lead for the development of a new class of antibiotics.
期刊介绍:
The Journal of Medicinal Chemistry is a prestigious biweekly peer-reviewed publication that focuses on the multifaceted field of medicinal chemistry. Since its inception in 1959 as the Journal of Medicinal and Pharmaceutical Chemistry, it has evolved to become a cornerstone in the dissemination of research findings related to the design, synthesis, and development of therapeutic agents.
The Journal of Medicinal Chemistry is recognized for its significant impact in the scientific community, as evidenced by its 2022 impact factor of 7.3. This metric reflects the journal's influence and the importance of its content in shaping the future of drug discovery and development. The journal serves as a vital resource for chemists, pharmacologists, and other researchers interested in the molecular mechanisms of drug action and the optimization of therapeutic compounds.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
