Zhe Li, Shunzhi Wang, Una Nattermann, Asim K. Bera, Andrew J. Borst, Muammer Y. Yaman, Matthew J. Bick, Erin C. Yang, William Sheffler, Byeongdu Lee, Soenke Seifert, Greg L. Hura, Hannah Nguyen, Alex Kang, Radhika Dalal, Joshua M. Lubner, Yang Hsia, Hugh Haddox, Alexis Courbet, Quinton Dowling, Marcos Miranda, Andrew Favor, Ali Etemadi, Natasha I. Edman, Wei Yang, Connor Weidle, Banumathi Sankaran, Babak Negahdari, Michael B. Ross, David S. Ginger, David Baker
{"title":"Accurate computational design of three-dimensional protein crystals","authors":"Zhe Li, Shunzhi Wang, Una Nattermann, Asim K. Bera, Andrew J. Borst, Muammer Y. Yaman, Matthew J. Bick, Erin C. Yang, William Sheffler, Byeongdu Lee, Soenke Seifert, Greg L. Hura, Hannah Nguyen, Alex Kang, Radhika Dalal, Joshua M. Lubner, Yang Hsia, Hugh Haddox, Alexis Courbet, Quinton Dowling, Marcos Miranda, Andrew Favor, Ali Etemadi, Natasha I. Edman, Wei Yang, Connor Weidle, Banumathi Sankaran, Babak Negahdari, Michael B. Ross, David S. Ginger, David Baker","doi":"10.1038/s41563-023-01683-1","DOIUrl":null,"url":null,"abstract":"Protein crystallization plays a central role in structural biology. Despite this, the process of crystallization remains poorly understood and highly empirical, with crystal contacts, lattice packing arrangements and space group preferences being largely unpredictable. Programming protein crystallization through precisely engineered side-chain–side-chain interactions across protein–protein interfaces is an outstanding challenge. Here we develop a general computational approach for designing three-dimensional protein crystals with prespecified lattice architectures at atomic accuracy that hierarchically constrains the overall number of degrees of freedom of the system. We design three pairs of oligomers that can be individually purified, and upon mixing, spontaneously self-assemble into >100 µm three-dimensional crystals. The structures of these crystals are nearly identical to the computational design models, closely corresponding in both overall architecture and the specific protein–protein interactions. The dimensions of the crystal unit cell can be systematically redesigned while retaining the space group symmetry and overall architecture, and the crystals are extremely porous and highly stable. Our approach enables the computational design of protein crystals with high accuracy, and the designed protein crystals, which have both structural and assembly information encoded in their primary sequences, provide a powerful platform for biological materials engineering. The process of protein crystallization is poorly understood and difficult to program through the primary sequence. Here the authors develop a computational approach to designing three-dimensional protein crystals with prespecified lattice architectures with high accuracy.","PeriodicalId":19058,"journal":{"name":"Nature Materials","volume":"22 12","pages":"1556-1563"},"PeriodicalIF":38.5000,"publicationDate":"2023-10-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Materials","FirstCategoryId":"88","ListUrlMain":"https://www.nature.com/articles/s41563-023-01683-1","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
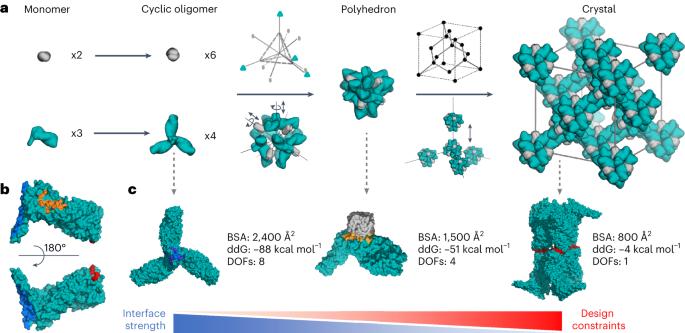
Protein crystallization plays a central role in structural biology. Despite this, the process of crystallization remains poorly understood and highly empirical, with crystal contacts, lattice packing arrangements and space group preferences being largely unpredictable. Programming protein crystallization through precisely engineered side-chain–side-chain interactions across protein–protein interfaces is an outstanding challenge. Here we develop a general computational approach for designing three-dimensional protein crystals with prespecified lattice architectures at atomic accuracy that hierarchically constrains the overall number of degrees of freedom of the system. We design three pairs of oligomers that can be individually purified, and upon mixing, spontaneously self-assemble into >100 µm three-dimensional crystals. The structures of these crystals are nearly identical to the computational design models, closely corresponding in both overall architecture and the specific protein–protein interactions. The dimensions of the crystal unit cell can be systematically redesigned while retaining the space group symmetry and overall architecture, and the crystals are extremely porous and highly stable. Our approach enables the computational design of protein crystals with high accuracy, and the designed protein crystals, which have both structural and assembly information encoded in their primary sequences, provide a powerful platform for biological materials engineering. The process of protein crystallization is poorly understood and difficult to program through the primary sequence. Here the authors develop a computational approach to designing three-dimensional protein crystals with prespecified lattice architectures with high accuracy.
期刊介绍:
Nature Materials is a monthly multi-disciplinary journal aimed at bringing together cutting-edge research across the entire spectrum of materials science and engineering. It covers all applied and fundamental aspects of the synthesis/processing, structure/composition, properties, and performance of materials. The journal recognizes that materials research has an increasing impact on classical disciplines such as physics, chemistry, and biology.
Additionally, Nature Materials provides a forum for the development of a common identity among materials scientists and encourages interdisciplinary collaboration. It takes an integrated and balanced approach to all areas of materials research, fostering the exchange of ideas between scientists involved in different disciplines.
Nature Materials is an invaluable resource for scientists in academia and industry who are active in discovering and developing materials and materials-related concepts. It offers engaging and informative papers of exceptional significance and quality, with the aim of influencing the development of society in the future.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
