{"title":"A unified molecular-wide and electron density based concept of chemical bonding","authors":"Ignacy Cukrowski","doi":"10.1002/wcms.1579","DOIUrl":null,"url":null,"abstract":"<p>Chemical bonding is at heart but, not being a quantum mechanical-defined physical property of a system, is a subject of endless and often fruitless debates. Having so many and very different models of chemical bonding without knowing what this really is does not make it easier. There is, however, a general agreement that concentrating electron density (ED) in and delocalizing ED to internuclear region is always associated with minimizing system's energy and synonymous with chemical bonding. Fragment, atomic, localized, delocalized, and interatomic (FALDI)-based density analysis involves entire space occupied by a molecule. From this molecular-wide and density-based methodology, it is possible to quantify localized and delocalized by all atoms ED at any coordinate <b>r</b>, including critical points on Bader's molecular graphs. Each atom and atom-pair contributions of delocalized density are quantified to reveal major players in the all-atom and molecular-wide chemical bonding. Partitioning the total ED to individual molecular or natural orbital's contributions using MO-ED and MO-DI methods, in conjunction with one dimensional (1D) cross section methodology, generates an orbital-based molecular-wide picture. This provides, besides reproducing results from FALDI, qualitative description of orbitals' nature that correlates well with classical understanding of bonding, nonbonding, and antibonding orbitals. A qualitative and quantitative impact of an immediate, distant, or molecular-wide molecular environment on intra- and intermolecular di-atomic, intra- and interfragment interactions is the domain of the Fragment Attributed Molecular System Energy Change (FAMSEC) family of methods. The FALDI, FAMSEC, MO-ED, MO-DI, and 1D cross section methodologies provide consistent and quantifiable physics-based picture of molecular-wide chemical bonding without invoking unicorns, such as a chemical bond.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"12 3","pages":""},"PeriodicalIF":27.0000,"publicationDate":"2021-10-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1579","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1579","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 4
Abstract
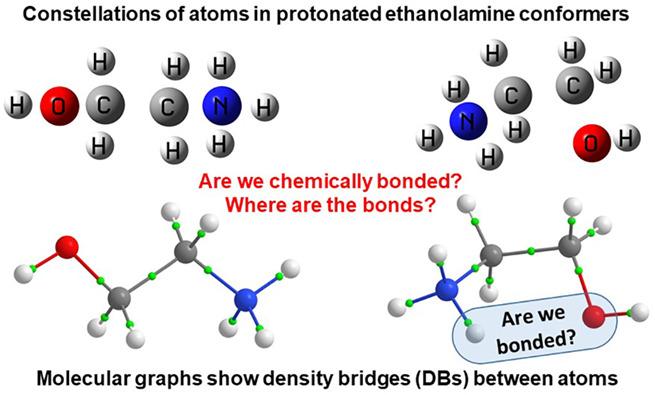
Chemical bonding is at heart but, not being a quantum mechanical-defined physical property of a system, is a subject of endless and often fruitless debates. Having so many and very different models of chemical bonding without knowing what this really is does not make it easier. There is, however, a general agreement that concentrating electron density (ED) in and delocalizing ED to internuclear region is always associated with minimizing system's energy and synonymous with chemical bonding. Fragment, atomic, localized, delocalized, and interatomic (FALDI)-based density analysis involves entire space occupied by a molecule. From this molecular-wide and density-based methodology, it is possible to quantify localized and delocalized by all atoms ED at any coordinate r, including critical points on Bader's molecular graphs. Each atom and atom-pair contributions of delocalized density are quantified to reveal major players in the all-atom and molecular-wide chemical bonding. Partitioning the total ED to individual molecular or natural orbital's contributions using MO-ED and MO-DI methods, in conjunction with one dimensional (1D) cross section methodology, generates an orbital-based molecular-wide picture. This provides, besides reproducing results from FALDI, qualitative description of orbitals' nature that correlates well with classical understanding of bonding, nonbonding, and antibonding orbitals. A qualitative and quantitative impact of an immediate, distant, or molecular-wide molecular environment on intra- and intermolecular di-atomic, intra- and interfragment interactions is the domain of the Fragment Attributed Molecular System Energy Change (FAMSEC) family of methods. The FALDI, FAMSEC, MO-ED, MO-DI, and 1D cross section methodologies provide consistent and quantifiable physics-based picture of molecular-wide chemical bonding without invoking unicorns, such as a chemical bond.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
