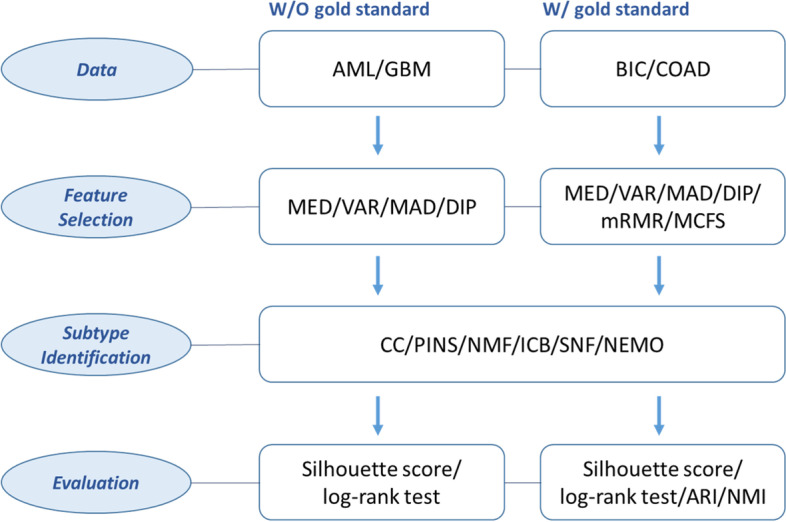
{"title":"Comparison of cancer subtype identification methods combined with feature selection methods in omics data analysis.","authors":"JiYoon Park, Jae Won Lee, Mira Park","doi":"10.1186/s13040-023-00334-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Cancer subtype identification is important for the early diagnosis of cancer and the provision of adequate treatment. Prior to identifying the subtype of cancer in a patient, feature selection is also crucial for reducing the dimensionality of the data by detecting genes that contain important information about the cancer subtype. Numerous cancer subtyping methods have been developed, and their performance has been compared. However, combinations of feature selection and subtype identification methods have rarely been considered. This study aimed to identify the best combination of variable selection and subtype identification methods in single omics data analysis.</p><p><strong>Results: </strong>Combinations of six filter-based methods and six unsupervised subtype identification methods were investigated using The Cancer Genome Atlas (TCGA) datasets for four cancers. The number of features selected varied, and several evaluation metrics were used. Although no single combination was found to have a distinctively good performance, Consensus Clustering (CC) and Neighborhood-Based Multi-omics Clustering (NEMO) used with variance-based feature selection had a tendency to show lower p-values, and nonnegative matrix factorization (NMF) stably showed good performance in many cases unless the Dip test was used for feature selection. In terms of accuracy, the combination of NMF and similarity network fusion (SNF) with Monte Carlo Feature Selection (MCFS) and Minimum-Redundancy Maximum Relevance (mRMR) showed good overall performance. NMF always showed among the worst performances without feature selection in all datasets, but performed much better when used with various feature selection methods. iClusterBayes (ICB) had decent performance when used without feature selection.</p><p><strong>Conclusions: </strong>Rather than a single method clearly emerging as optimal, the best methodology was different depending on the data used, the number of features selected, and the evaluation method. A guideline for choosing the best combination method under various situations is provided.</p>","PeriodicalId":48947,"journal":{"name":"Biodata Mining","volume":"16 1","pages":"18"},"PeriodicalIF":6.1000,"publicationDate":"2023-07-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10329370/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biodata Mining","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13040-023-00334-0","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Cancer subtype identification is important for the early diagnosis of cancer and the provision of adequate treatment. Prior to identifying the subtype of cancer in a patient, feature selection is also crucial for reducing the dimensionality of the data by detecting genes that contain important information about the cancer subtype. Numerous cancer subtyping methods have been developed, and their performance has been compared. However, combinations of feature selection and subtype identification methods have rarely been considered. This study aimed to identify the best combination of variable selection and subtype identification methods in single omics data analysis.
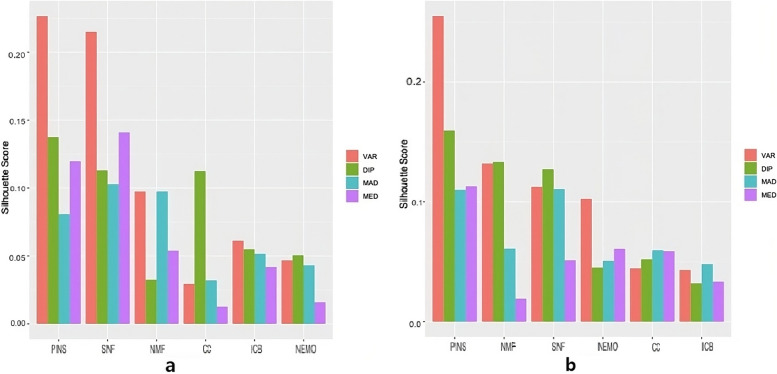
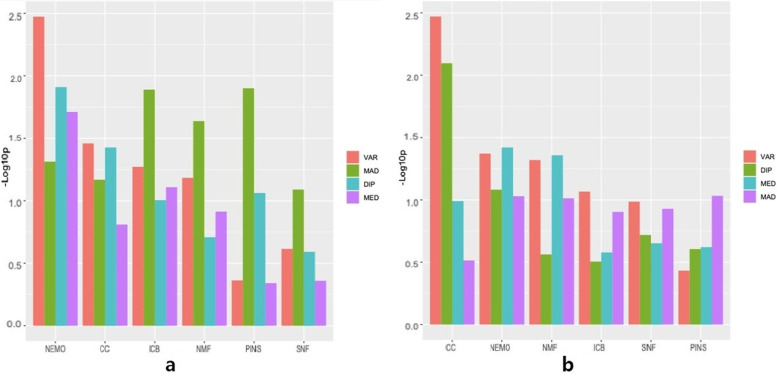
Results: Combinations of six filter-based methods and six unsupervised subtype identification methods were investigated using The Cancer Genome Atlas (TCGA) datasets for four cancers. The number of features selected varied, and several evaluation metrics were used. Although no single combination was found to have a distinctively good performance, Consensus Clustering (CC) and Neighborhood-Based Multi-omics Clustering (NEMO) used with variance-based feature selection had a tendency to show lower p-values, and nonnegative matrix factorization (NMF) stably showed good performance in many cases unless the Dip test was used for feature selection. In terms of accuracy, the combination of NMF and similarity network fusion (SNF) with Monte Carlo Feature Selection (MCFS) and Minimum-Redundancy Maximum Relevance (mRMR) showed good overall performance. NMF always showed among the worst performances without feature selection in all datasets, but performed much better when used with various feature selection methods. iClusterBayes (ICB) had decent performance when used without feature selection.
Conclusions: Rather than a single method clearly emerging as optimal, the best methodology was different depending on the data used, the number of features selected, and the evaluation method. A guideline for choosing the best combination method under various situations is provided.
期刊介绍:
BioData Mining is an open access, open peer-reviewed journal encompassing research on all aspects of data mining applied to high-dimensional biological and biomedical data, focusing on computational aspects of knowledge discovery from large-scale genetic, transcriptomic, genomic, proteomic, and metabolomic data.
Topical areas include, but are not limited to:
-Development, evaluation, and application of novel data mining and machine learning algorithms.
-Adaptation, evaluation, and application of traditional data mining and machine learning algorithms.
-Open-source software for the application of data mining and machine learning algorithms.
-Design, development and integration of databases, software and web services for the storage, management, retrieval, and analysis of data from large scale studies.
-Pre-processing, post-processing, modeling, and interpretation of data mining and machine learning results for biological interpretation and knowledge discovery.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
