Pavel V Afonine, Oleg V Sobolev, Nigel W Moriarty, Thomas C Terwilliger, Paul D Adams
{"title":"Overall protein structure quality assessment using hydrogen-bonding parameters.","authors":"Pavel V Afonine, Oleg V Sobolev, Nigel W Moriarty, Thomas C Terwilliger, Paul D Adams","doi":"10.1107/S2059798323005077","DOIUrl":null,"url":null,"abstract":"<p><p>Atomic model refinement at low resolution is often a challenging task. This is mostly because the experimental data are not sufficiently detailed to be described by atomic models. To make refinement practical and ensure that a refined atomic model is geometrically meaningful, additional information needs to be used such as restraints on Ramachandran plot distributions or residue side-chain rotameric states. However, using Ramachandran plots or rotameric states as refinement targets diminishes the validating power of these tools. Therefore, finding additional model-validation criteria that are not used or are difficult to use as refinement goals is desirable. Hydrogen bonds are one of the important noncovalent interactions that shape and maintain protein structure. These interactions can be characterized by a specific geometry of hydrogen donor and acceptor atoms. Systematic analysis of these geometries performed for quality-filtered high-resolution models of proteins from the Protein Data Bank shows that they have a distinct and a conserved distribution. Here, it is demonstrated how this information can be used for atomic model validation.</p>","PeriodicalId":7116,"journal":{"name":"Acta Crystallographica. Section D, Structural Biology","volume":"79 Pt 8","pages":"684-693"},"PeriodicalIF":3.8000,"publicationDate":"2023-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10394671/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Crystallographica. Section D, Structural Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1107/S2059798323005077","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/7/11 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
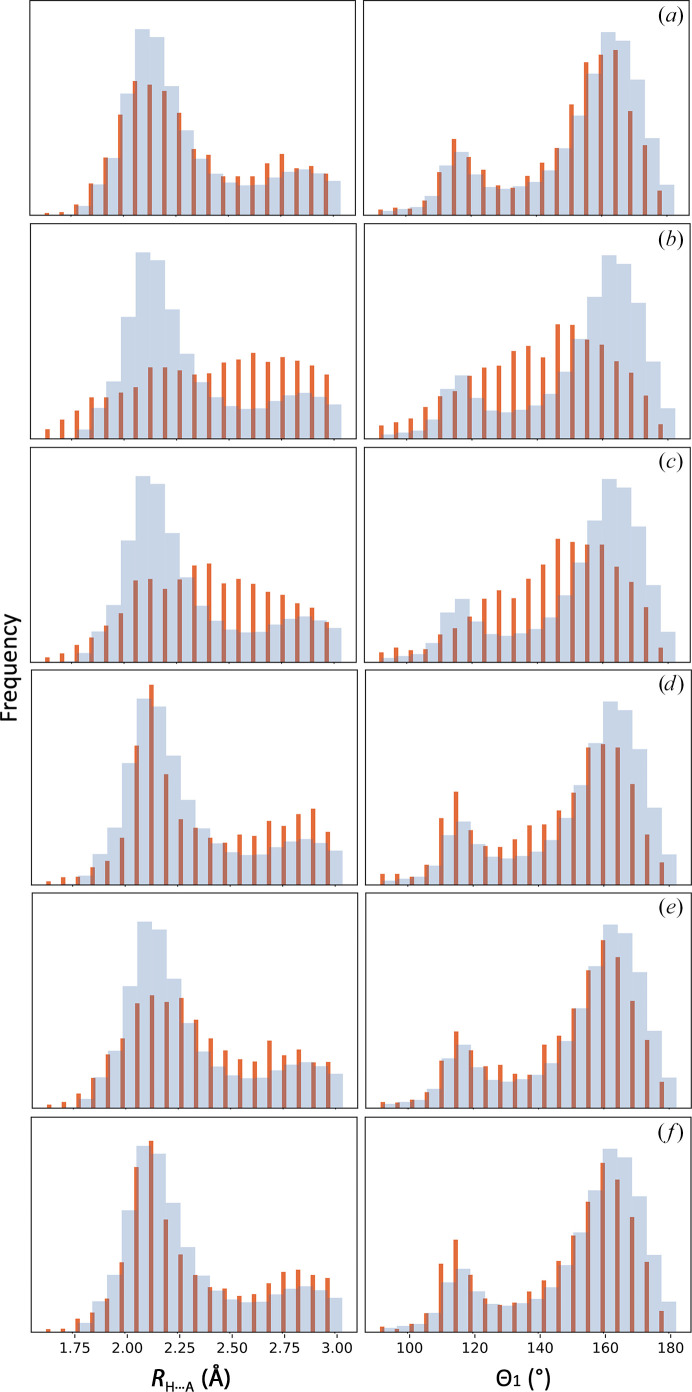
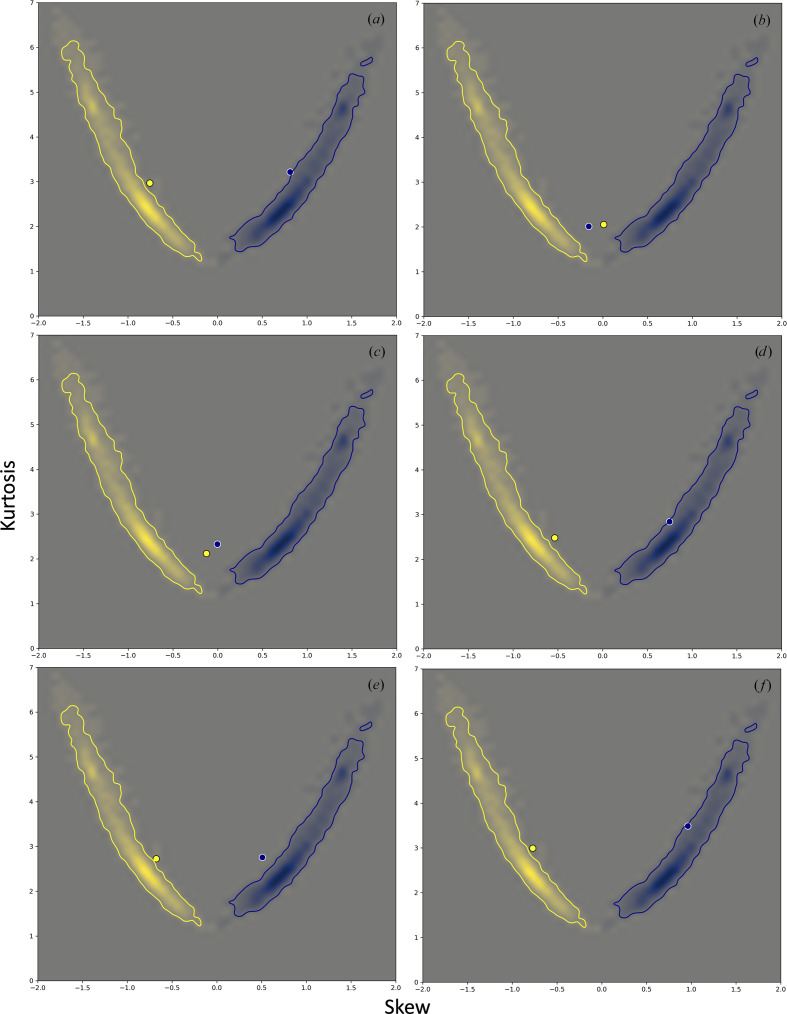
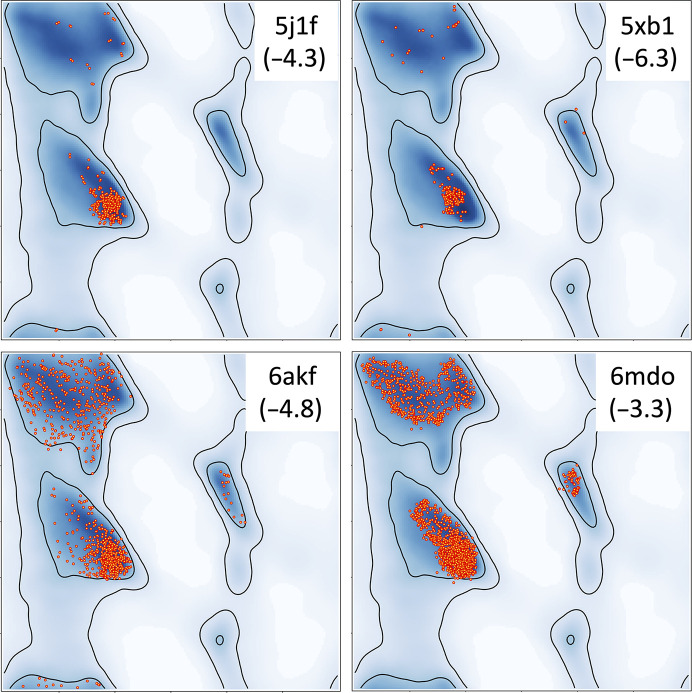
Atomic model refinement at low resolution is often a challenging task. This is mostly because the experimental data are not sufficiently detailed to be described by atomic models. To make refinement practical and ensure that a refined atomic model is geometrically meaningful, additional information needs to be used such as restraints on Ramachandran plot distributions or residue side-chain rotameric states. However, using Ramachandran plots or rotameric states as refinement targets diminishes the validating power of these tools. Therefore, finding additional model-validation criteria that are not used or are difficult to use as refinement goals is desirable. Hydrogen bonds are one of the important noncovalent interactions that shape and maintain protein structure. These interactions can be characterized by a specific geometry of hydrogen donor and acceptor atoms. Systematic analysis of these geometries performed for quality-filtered high-resolution models of proteins from the Protein Data Bank shows that they have a distinct and a conserved distribution. Here, it is demonstrated how this information can be used for atomic model validation.
期刊介绍:
Acta Crystallographica Section D welcomes the submission of articles covering any aspect of structural biology, with a particular emphasis on the structures of biological macromolecules or the methods used to determine them.
Reports on new structures of biological importance may address the smallest macromolecules to the largest complex molecular machines. These structures may have been determined using any structural biology technique including crystallography, NMR, cryoEM and/or other techniques. The key criterion is that such articles must present significant new insights into biological, chemical or medical sciences. The inclusion of complementary data that support the conclusions drawn from the structural studies (such as binding studies, mass spectrometry, enzyme assays, or analysis of mutants or other modified forms of biological macromolecule) is encouraged.
Methods articles may include new approaches to any aspect of biological structure determination or structure analysis but will only be accepted where they focus on new methods that are demonstrated to be of general applicability and importance to structural biology. Articles describing particularly difficult problems in structural biology are also welcomed, if the analysis would provide useful insights to others facing similar problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
