{"title":"gmXtal:用 GROMACS 烹饪晶体。","authors":"Pavel Buslaev, Gerrit Groenhof","doi":"10.1007/s10930-023-10141-5","DOIUrl":null,"url":null,"abstract":"<div><p>Molecular dynamics (MD) simulations are routinely performed of biomolecules in solution, because this is their native environment. However, the structures used in such simulations are often obtained with X-ray crystallography, which provides the atomic coordinates of the biomolecule in a crystal environment. With the advent of free electron lasers and time-resolved techniques, X-ray crystallography can now also access metastable states that are intermediates in a biochemical process. Such experiments provide additional data, which can be used, for example, to optimize MD force fields. Doing so requires that the simulation of the biomolecule is also performed in the crystal environment. However, in contrast to simulations of biomolecules in solution, setting up a crystal is challenging. In particular, because not all solvent molecules are resolved in X-ray crystallography, adding a suitable number of solvent molecules, such that the properties of the crystallographic unit cell are preserved in the simulation, can be difficult and typically is a trial-and-error based procedure requiring manual interventions. Such interventions preclude high throughput applications. To overcome this bottleneck, we introduce <b>gmXtal</b>, a tool for setting up crystal simulations for MD simulations with GROMACS. With the information from the protein data bank (rcsb.org) <b>gmXtal</b> automatically (i) builds the crystallographic unit cell; (ii) sets the protonation of titratable residues; (iii) builds missing residues that were not resolved experimentally; and (iv) adds an appropriate number of solvent molecules to the system. <b>gmXtal</b> is available as a standalone tool https://gitlab.com/pbuslaev/gmxtal.</p><h3>Graphical Abstract</h3>\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":793,"journal":{"name":"The Protein Journal","volume":"43 2","pages":"200 - 206"},"PeriodicalIF":1.9000,"publicationDate":"2023-08-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11058868/pdf/","citationCount":"0","resultStr":"{\"title\":\"gmXtal: Cooking Crystals with GROMACS\",\"authors\":\"Pavel Buslaev, Gerrit Groenhof\",\"doi\":\"10.1007/s10930-023-10141-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Molecular dynamics (MD) simulations are routinely performed of biomolecules in solution, because this is their native environment. However, the structures used in such simulations are often obtained with X-ray crystallography, which provides the atomic coordinates of the biomolecule in a crystal environment. With the advent of free electron lasers and time-resolved techniques, X-ray crystallography can now also access metastable states that are intermediates in a biochemical process. Such experiments provide additional data, which can be used, for example, to optimize MD force fields. Doing so requires that the simulation of the biomolecule is also performed in the crystal environment. However, in contrast to simulations of biomolecules in solution, setting up a crystal is challenging. In particular, because not all solvent molecules are resolved in X-ray crystallography, adding a suitable number of solvent molecules, such that the properties of the crystallographic unit cell are preserved in the simulation, can be difficult and typically is a trial-and-error based procedure requiring manual interventions. Such interventions preclude high throughput applications. To overcome this bottleneck, we introduce <b>gmXtal</b>, a tool for setting up crystal simulations for MD simulations with GROMACS. With the information from the protein data bank (rcsb.org) <b>gmXtal</b> automatically (i) builds the crystallographic unit cell; (ii) sets the protonation of titratable residues; (iii) builds missing residues that were not resolved experimentally; and (iv) adds an appropriate number of solvent molecules to the system. <b>gmXtal</b> is available as a standalone tool https://gitlab.com/pbuslaev/gmxtal.</p><h3>Graphical Abstract</h3>\\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>\",\"PeriodicalId\":793,\"journal\":{\"name\":\"The Protein Journal\",\"volume\":\"43 2\",\"pages\":\"200 - 206\"},\"PeriodicalIF\":1.9000,\"publicationDate\":\"2023-08-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11058868/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Protein Journal\",\"FirstCategoryId\":\"2\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s10930-023-10141-5\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Protein Journal","FirstCategoryId":"2","ListUrlMain":"https://link.springer.com/article/10.1007/s10930-023-10141-5","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
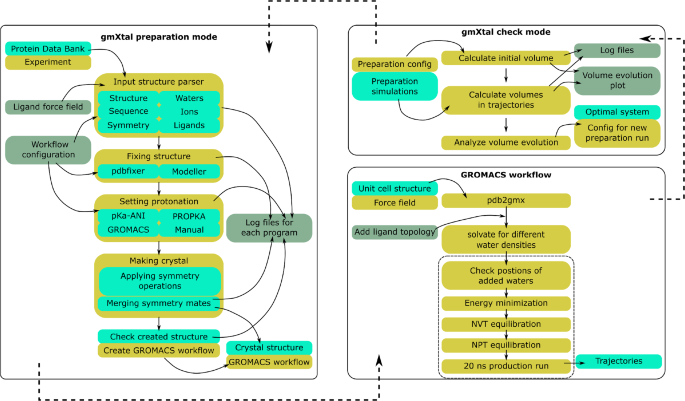
分子动力学(MD)模拟通常是对溶液中的生物分子进行模拟,因为这是生物分子的原生环境。然而,此类模拟中使用的结构通常是通过 X 射线晶体学获得的,X 射线晶体学提供了晶体环境中生物大分子的原子坐标。随着自由电子激光器和时间分辨技术的出现,X 射线晶体学现在还能获取作为生化过程中间产物的蜕变态。这些实验提供了额外的数据,可用于优化 MD 力场等。这样做需要在晶体环境中对生物分子进行模拟。然而,与溶液中的生物分子模拟相比,晶体的建立具有挑战性。特别是,由于并非所有的溶剂分子都能在 X 射线晶体学中解析,因此要添加适当数量的溶剂分子,从而在模拟中保留晶体学单元格的特性,可能会非常困难,而且通常是一个需要人工干预的试错过程。这种干预排除了高通量应用。为了克服这一瓶颈,我们推出了 gmXtal,这是一种利用 GROMACS 进行 MD 模拟的晶体模拟设置工具。利用蛋白质数据库(rcsb.org)中的信息,gmXtal 可自动:(i) 建立晶体学单元格;(ii) 设置可滴定残基的质子化;(iii) 建立实验中未解析的缺失残基;(iv) 为系统添加适当数量的溶剂分子。
Molecular dynamics (MD) simulations are routinely performed of biomolecules in solution, because this is their native environment. However, the structures used in such simulations are often obtained with X-ray crystallography, which provides the atomic coordinates of the biomolecule in a crystal environment. With the advent of free electron lasers and time-resolved techniques, X-ray crystallography can now also access metastable states that are intermediates in a biochemical process. Such experiments provide additional data, which can be used, for example, to optimize MD force fields. Doing so requires that the simulation of the biomolecule is also performed in the crystal environment. However, in contrast to simulations of biomolecules in solution, setting up a crystal is challenging. In particular, because not all solvent molecules are resolved in X-ray crystallography, adding a suitable number of solvent molecules, such that the properties of the crystallographic unit cell are preserved in the simulation, can be difficult and typically is a trial-and-error based procedure requiring manual interventions. Such interventions preclude high throughput applications. To overcome this bottleneck, we introduce gmXtal, a tool for setting up crystal simulations for MD simulations with GROMACS. With the information from the protein data bank (rcsb.org) gmXtal automatically (i) builds the crystallographic unit cell; (ii) sets the protonation of titratable residues; (iii) builds missing residues that were not resolved experimentally; and (iv) adds an appropriate number of solvent molecules to the system. gmXtal is available as a standalone tool https://gitlab.com/pbuslaev/gmxtal.
期刊介绍:
The Protein Journal (formerly the Journal of Protein Chemistry) publishes original research work on all aspects of proteins and peptides. These include studies concerned with covalent or three-dimensional structure determination (X-ray, NMR, cryoEM, EPR/ESR, optical methods, etc.), computational aspects of protein structure and function, protein folding and misfolding, assembly, genetics, evolution, proteomics, molecular biology, protein engineering, protein nanotechnology, protein purification and analysis and peptide synthesis, as well as the elucidation and interpretation of the molecular bases of biological activities of proteins and peptides. We accept original research papers, reviews, mini-reviews, hypotheses, opinion papers, and letters to the editor.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
