{"title":"基于多组学生物学网络的前列腺癌驱动模块识别","authors":"Zhongli Chen, Biting Liang, Yingfu Wu, Haoru Zhou, Yuchen Wang, Hao Wu","doi":"10.1049/syb2.12050","DOIUrl":null,"url":null,"abstract":"<p>The development of sequencing technology has promoted the expansion of cancer genome data. It is necessary to identify the pathogenesis of cancer at the molecular level and explore reliable treatment methods and precise drug targets in cancer by identifying carcinogenic functional modules in massive multi-omics data. However, there are still limitations to identifying carcinogenic driver modules by utilising genetic characteristics simply. Therefore, this study proposes a computational method, NetAP, to identify driver modules in prostate cancer. Firstly, high mutual exclusivity, high coverage, and high topological similarity between genes are integrated to construct a weight function, which calculates the weight of gene pairs in a biological network. Secondly, the random walk method is utilised to reevaluate the strength of interaction among genes. Finally, the optimal driver modules are identified by utilising the affinity propagation algorithm. According to the results, the authors’ method identifies more validated driver genes and driver modules compared with the other previous methods. Thus, the proposed NetAP method can identify carcinogenic driver modules effectively and reliably, and the experimental results provide a powerful basis for cancer diagnosis, treatment and drug targets.</p>","PeriodicalId":50379,"journal":{"name":"IET Systems Biology","volume":"16 6","pages":"187-200"},"PeriodicalIF":1.9000,"publicationDate":"2022-08-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/e8/7f/SYB2-16-187.PMC9675413.pdf","citationCount":"1","resultStr":"{\"title\":\"Identifying driver modules based on multi-omics biological networks in prostate cancer\",\"authors\":\"Zhongli Chen, Biting Liang, Yingfu Wu, Haoru Zhou, Yuchen Wang, Hao Wu\",\"doi\":\"10.1049/syb2.12050\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The development of sequencing technology has promoted the expansion of cancer genome data. It is necessary to identify the pathogenesis of cancer at the molecular level and explore reliable treatment methods and precise drug targets in cancer by identifying carcinogenic functional modules in massive multi-omics data. However, there are still limitations to identifying carcinogenic driver modules by utilising genetic characteristics simply. Therefore, this study proposes a computational method, NetAP, to identify driver modules in prostate cancer. Firstly, high mutual exclusivity, high coverage, and high topological similarity between genes are integrated to construct a weight function, which calculates the weight of gene pairs in a biological network. Secondly, the random walk method is utilised to reevaluate the strength of interaction among genes. Finally, the optimal driver modules are identified by utilising the affinity propagation algorithm. According to the results, the authors’ method identifies more validated driver genes and driver modules compared with the other previous methods. Thus, the proposed NetAP method can identify carcinogenic driver modules effectively and reliably, and the experimental results provide a powerful basis for cancer diagnosis, treatment and drug targets.</p>\",\"PeriodicalId\":50379,\"journal\":{\"name\":\"IET Systems Biology\",\"volume\":\"16 6\",\"pages\":\"187-200\"},\"PeriodicalIF\":1.9000,\"publicationDate\":\"2022-08-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/e8/7f/SYB2-16-187.PMC9675413.pdf\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"IET Systems Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1049/syb2.12050\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"IET Systems Biology","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1049/syb2.12050","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
Identifying driver modules based on multi-omics biological networks in prostate cancer
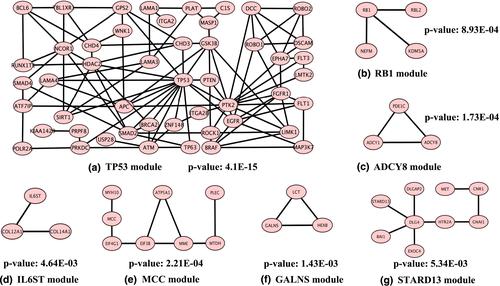
The development of sequencing technology has promoted the expansion of cancer genome data. It is necessary to identify the pathogenesis of cancer at the molecular level and explore reliable treatment methods and precise drug targets in cancer by identifying carcinogenic functional modules in massive multi-omics data. However, there are still limitations to identifying carcinogenic driver modules by utilising genetic characteristics simply. Therefore, this study proposes a computational method, NetAP, to identify driver modules in prostate cancer. Firstly, high mutual exclusivity, high coverage, and high topological similarity between genes are integrated to construct a weight function, which calculates the weight of gene pairs in a biological network. Secondly, the random walk method is utilised to reevaluate the strength of interaction among genes. Finally, the optimal driver modules are identified by utilising the affinity propagation algorithm. According to the results, the authors’ method identifies more validated driver genes and driver modules compared with the other previous methods. Thus, the proposed NetAP method can identify carcinogenic driver modules effectively and reliably, and the experimental results provide a powerful basis for cancer diagnosis, treatment and drug targets.
期刊介绍:
IET Systems Biology covers intra- and inter-cellular dynamics, using systems- and signal-oriented approaches. Papers that analyse genomic data in order to identify variables and basic relationships between them are considered if the results provide a basis for mathematical modelling and simulation of cellular dynamics. Manuscripts on molecular and cell biological studies are encouraged if the aim is a systems approach to dynamic interactions within and between cells.
The scope includes the following topics:
Genomics, transcriptomics, proteomics, metabolomics, cells, tissue and the physiome; molecular and cellular interaction, gene, cell and protein function; networks and pathways; metabolism and cell signalling; dynamics, regulation and control; systems, signals, and information; experimental data analysis; mathematical modelling, simulation and theoretical analysis; biological modelling, simulation, prediction and control; methodologies, databases, tools and algorithms for modelling and simulation; modelling, analysis and control of biological networks; synthetic biology and bioengineering based on systems biology.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
