Liwei Chang, Arup Mondal, Bhumika Singh, Yisel Martínez-Noa, Alberto Perez
{"title":"肽类药物发现的革命性变革:后阿尔法折叠时代的进步","authors":"Liwei Chang, Arup Mondal, Bhumika Singh, Yisel Martínez-Noa, Alberto Perez","doi":"10.1002/wcms.1693","DOIUrl":null,"url":null,"abstract":"<p>Peptide-based drugs offer high specificity, potency, and selectivity. However, their inherent flexibility and differences in conformational preferences between their free and bound states create unique challenges that have hindered progress in effective drug discovery pipelines. The emergence of AlphaFold (AF) and Artificial Intelligence (AI) presents new opportunities for enhancing peptide-based drug discovery. We explore recent advancements that facilitate a successful peptide drug discovery pipeline, considering peptides' attractive therapeutic properties and strategies to enhance their stability and bioavailability. AF enables efficient and accurate prediction of peptide-protein structures, addressing a critical requirement in computational drug discovery pipelines. In the post-AF era, we are witnessing rapid progress with the potential to revolutionize peptide-based drug discovery such as the ability to rank peptide binders or classify them as binders/non-binders and the ability to design novel peptide sequences. However, AI-based methods are struggling due to the lack of well-curated datasets, for example to accommodate modified amino acids or unconventional cyclization. Thus, physics-based methods, such as docking or molecular dynamics simulations, continue to hold a complementary role in peptide drug discovery pipelines. Moreover, MD-based tools offer valuable insights into binding mechanisms, as well as the thermodynamic and kinetic properties of complexes. As we navigate this evolving landscape, a synergistic integration of AI and physics-based methods holds the promise of reshaping the landscape of peptide-based drug discovery.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"14 1","pages":""},"PeriodicalIF":27.0000,"publicationDate":"2023-11-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Revolutionizing peptide-based drug discovery: Advances in the post-AlphaFold era\",\"authors\":\"Liwei Chang, Arup Mondal, Bhumika Singh, Yisel Martínez-Noa, Alberto Perez\",\"doi\":\"10.1002/wcms.1693\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Peptide-based drugs offer high specificity, potency, and selectivity. However, their inherent flexibility and differences in conformational preferences between their free and bound states create unique challenges that have hindered progress in effective drug discovery pipelines. The emergence of AlphaFold (AF) and Artificial Intelligence (AI) presents new opportunities for enhancing peptide-based drug discovery. We explore recent advancements that facilitate a successful peptide drug discovery pipeline, considering peptides' attractive therapeutic properties and strategies to enhance their stability and bioavailability. AF enables efficient and accurate prediction of peptide-protein structures, addressing a critical requirement in computational drug discovery pipelines. In the post-AF era, we are witnessing rapid progress with the potential to revolutionize peptide-based drug discovery such as the ability to rank peptide binders or classify them as binders/non-binders and the ability to design novel peptide sequences. However, AI-based methods are struggling due to the lack of well-curated datasets, for example to accommodate modified amino acids or unconventional cyclization. Thus, physics-based methods, such as docking or molecular dynamics simulations, continue to hold a complementary role in peptide drug discovery pipelines. Moreover, MD-based tools offer valuable insights into binding mechanisms, as well as the thermodynamic and kinetic properties of complexes. As we navigate this evolving landscape, a synergistic integration of AI and physics-based methods holds the promise of reshaping the landscape of peptide-based drug discovery.</p><p>This article is categorized under:\\n </p>\",\"PeriodicalId\":236,\"journal\":{\"name\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"volume\":\"14 1\",\"pages\":\"\"},\"PeriodicalIF\":27.0000,\"publicationDate\":\"2023-11-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://wires.onlinelibrary.wiley.com/doi/10.1002/wcms.1693\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://wires.onlinelibrary.wiley.com/doi/10.1002/wcms.1693","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
摘要
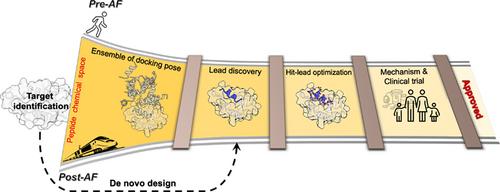
肽类药物具有高特异性、高效力和高选择性。然而,多肽固有的灵活性以及游离态和结合态之间构象偏好的差异带来了独特的挑战,阻碍了有效药物发现管道的进展。阿尔法折叠(AlphaFold,AF)和人工智能(Artificial Intelligence,AI)的出现为加强基于多肽的药物发现带来了新的机遇。考虑到多肽极具吸引力的治疗特性以及提高其稳定性和生物利用度的策略,我们将探讨促进多肽药物研发管道取得成功的最新进展。AF 能够高效、准确地预测多肽-蛋白质结构,满足了计算药物发现管道的关键要求。在后 AF 时代,我们目睹了快速的进步,这些进步有可能彻底改变基于多肽的药物发现,例如对多肽结合体进行排序或将其分类为结合体/非结合体的能力,以及设计新型多肽序列的能力。然而,基于人工智能的方法由于缺乏完善的数据集而举步维艰,例如,无法适应修饰氨基酸或非常规环化。因此,基于物理的方法,如对接或分子动力学模拟,在多肽药物发现管道中仍起着补充作用。此外,基于 MD 的工具还能提供有关结合机制以及复合物热力学和动力学特性的宝贵见解。在我们驾驭这种不断变化的格局时,人工智能和基于物理学的方法的协同整合有望重塑多肽药物发现的格局:
Revolutionizing peptide-based drug discovery: Advances in the post-AlphaFold era
Peptide-based drugs offer high specificity, potency, and selectivity. However, their inherent flexibility and differences in conformational preferences between their free and bound states create unique challenges that have hindered progress in effective drug discovery pipelines. The emergence of AlphaFold (AF) and Artificial Intelligence (AI) presents new opportunities for enhancing peptide-based drug discovery. We explore recent advancements that facilitate a successful peptide drug discovery pipeline, considering peptides' attractive therapeutic properties and strategies to enhance their stability and bioavailability. AF enables efficient and accurate prediction of peptide-protein structures, addressing a critical requirement in computational drug discovery pipelines. In the post-AF era, we are witnessing rapid progress with the potential to revolutionize peptide-based drug discovery such as the ability to rank peptide binders or classify them as binders/non-binders and the ability to design novel peptide sequences. However, AI-based methods are struggling due to the lack of well-curated datasets, for example to accommodate modified amino acids or unconventional cyclization. Thus, physics-based methods, such as docking or molecular dynamics simulations, continue to hold a complementary role in peptide drug discovery pipelines. Moreover, MD-based tools offer valuable insights into binding mechanisms, as well as the thermodynamic and kinetic properties of complexes. As we navigate this evolving landscape, a synergistic integration of AI and physics-based methods holds the promise of reshaping the landscape of peptide-based drug discovery.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
