Sarah Löffelsender, Pierre Beaujean, Marc de Wergifosse
{"title":"评估大型系统非线性光学特性的简化量子化学方法","authors":"Sarah Löffelsender, Pierre Beaujean, Marc de Wergifosse","doi":"10.1002/wcms.1695","DOIUrl":null,"url":null,"abstract":"<p>This review presents the theoretical background concerning simplified quantum chemistry (sQC) methods to compute non-linear optical (NLO) properties and their applications to large systems. To evaluate any NLO responses such as hyperpolarizabilities or two-photon absorption (2PA), one should evidently perform first a ground state calculation and compute its response. Because of this, methods used to compute ground states of large systems are outlined, especially the xTB (extended tight-binding) scheme. An overview on approaches to compute excited state and response properties is given, emphasizing the simplified time-dependent density functional theory (sTD-DFT). The formalism of the eXact integral sTD-DFT (XsTD-DFT) method is also introduced. For the first hyperpolarizability, 2PA, excited state absorption, and second hyperpolarizability, a brief historical review is given on early-stage semi-empirical method applications to systems that were considered large at the time. Then, we showcase recent applications with sQC methods, especially the sTD-DFT scheme to large challenging systems such as fluorescent proteins or fluorescent organic nanoparticles as well as dynamic structural effects on flexible tryptophan-rich peptides and gramicidin A. Thanks to the sTD-DFT-xTB scheme, all-atom quantum chemistry methodologies are now possible for the computation of the first hyperpolarizability and 2PA of systems up to 5000 atoms. This review concludes by summing-up current and future method developments in the sQC framework as well as forthcoming applications on large systems.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"14 1","pages":""},"PeriodicalIF":27.0000,"publicationDate":"2023-11-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Simplified quantum chemistry methods to evaluate non-linear optical properties of large systems\",\"authors\":\"Sarah Löffelsender, Pierre Beaujean, Marc de Wergifosse\",\"doi\":\"10.1002/wcms.1695\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>This review presents the theoretical background concerning simplified quantum chemistry (sQC) methods to compute non-linear optical (NLO) properties and their applications to large systems. To evaluate any NLO responses such as hyperpolarizabilities or two-photon absorption (2PA), one should evidently perform first a ground state calculation and compute its response. Because of this, methods used to compute ground states of large systems are outlined, especially the xTB (extended tight-binding) scheme. An overview on approaches to compute excited state and response properties is given, emphasizing the simplified time-dependent density functional theory (sTD-DFT). The formalism of the eXact integral sTD-DFT (XsTD-DFT) method is also introduced. For the first hyperpolarizability, 2PA, excited state absorption, and second hyperpolarizability, a brief historical review is given on early-stage semi-empirical method applications to systems that were considered large at the time. Then, we showcase recent applications with sQC methods, especially the sTD-DFT scheme to large challenging systems such as fluorescent proteins or fluorescent organic nanoparticles as well as dynamic structural effects on flexible tryptophan-rich peptides and gramicidin A. Thanks to the sTD-DFT-xTB scheme, all-atom quantum chemistry methodologies are now possible for the computation of the first hyperpolarizability and 2PA of systems up to 5000 atoms. This review concludes by summing-up current and future method developments in the sQC framework as well as forthcoming applications on large systems.</p><p>This article is categorized under:\\n </p>\",\"PeriodicalId\":236,\"journal\":{\"name\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"volume\":\"14 1\",\"pages\":\"\"},\"PeriodicalIF\":27.0000,\"publicationDate\":\"2023-11-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://wires.onlinelibrary.wiley.com/doi/10.1002/wcms.1695\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://wires.onlinelibrary.wiley.com/doi/10.1002/wcms.1695","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Simplified quantum chemistry methods to evaluate non-linear optical properties of large systems
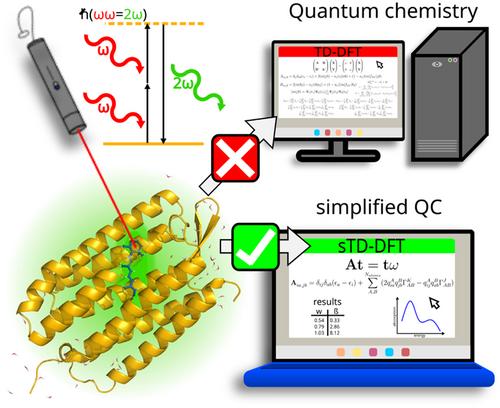
This review presents the theoretical background concerning simplified quantum chemistry (sQC) methods to compute non-linear optical (NLO) properties and their applications to large systems. To evaluate any NLO responses such as hyperpolarizabilities or two-photon absorption (2PA), one should evidently perform first a ground state calculation and compute its response. Because of this, methods used to compute ground states of large systems are outlined, especially the xTB (extended tight-binding) scheme. An overview on approaches to compute excited state and response properties is given, emphasizing the simplified time-dependent density functional theory (sTD-DFT). The formalism of the eXact integral sTD-DFT (XsTD-DFT) method is also introduced. For the first hyperpolarizability, 2PA, excited state absorption, and second hyperpolarizability, a brief historical review is given on early-stage semi-empirical method applications to systems that were considered large at the time. Then, we showcase recent applications with sQC methods, especially the sTD-DFT scheme to large challenging systems such as fluorescent proteins or fluorescent organic nanoparticles as well as dynamic structural effects on flexible tryptophan-rich peptides and gramicidin A. Thanks to the sTD-DFT-xTB scheme, all-atom quantum chemistry methodologies are now possible for the computation of the first hyperpolarizability and 2PA of systems up to 5000 atoms. This review concludes by summing-up current and future method developments in the sQC framework as well as forthcoming applications on large systems.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
