{"title":"二氧化碳在 δ-Pu(1 0 0) 表面上的反应机理:DFT 研究","authors":"Jintao Wang, Xin Qu, Haijian Xue, Guiyun Hang, Tao Wang, Wenli Yu","doi":"10.1016/j.commatsci.2024.113267","DOIUrl":null,"url":null,"abstract":"<div><p>This investigation elucidates the adsorptive and dissociative interactions of CO<sub>2</sub> and CO molecules with the δ-Pu (1<!--> <!-->0<!--> <!-->0) surface, utilizing state-of-the-art first-principles calculations. The study unearths that the hollow site epitomizes the most favorable adsorption locus for both CO<sub>2</sub> and CO. Within the paramount adsorption configuration, there prevails a propensity for both species to partake in dissociative adsorption, characterized by CO<sub>2</sub> cleaving into an O atom and a concomitant CO moiety, whereas CO disintegrates into discrete C and O atoms. The dissociation energy barriers conducive to such configurations are computed to be 3.0568 eV for CO<sub>2</sub> and 5.1667 eV for CO. A thorough analysis of electronic charges and density of states intimates a transfer of electrons from the Plutonium surface atoms to the carbonaceous adsorbates during dissociation. Post dissociation of the C-O bond, the 6<em>d</em> orbitals of Plutonium engage in electronic hybridization with the 2<em>p</em> orbitals of Carbon, culminating in the constitution of ionic bonding.</p></div>","PeriodicalId":10650,"journal":{"name":"Computational Materials Science","volume":"244 ","pages":"Article 113267"},"PeriodicalIF":3.3000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Reaction mechanism of CO2 on the surface δ-Pu(1 0 0):a DFT study\",\"authors\":\"Jintao Wang, Xin Qu, Haijian Xue, Guiyun Hang, Tao Wang, Wenli Yu\",\"doi\":\"10.1016/j.commatsci.2024.113267\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>This investigation elucidates the adsorptive and dissociative interactions of CO<sub>2</sub> and CO molecules with the δ-Pu (1<!--> <!-->0<!--> <!-->0) surface, utilizing state-of-the-art first-principles calculations. The study unearths that the hollow site epitomizes the most favorable adsorption locus for both CO<sub>2</sub> and CO. Within the paramount adsorption configuration, there prevails a propensity for both species to partake in dissociative adsorption, characterized by CO<sub>2</sub> cleaving into an O atom and a concomitant CO moiety, whereas CO disintegrates into discrete C and O atoms. The dissociation energy barriers conducive to such configurations are computed to be 3.0568 eV for CO<sub>2</sub> and 5.1667 eV for CO. A thorough analysis of electronic charges and density of states intimates a transfer of electrons from the Plutonium surface atoms to the carbonaceous adsorbates during dissociation. Post dissociation of the C-O bond, the 6<em>d</em> orbitals of Plutonium engage in electronic hybridization with the 2<em>p</em> orbitals of Carbon, culminating in the constitution of ionic bonding.</p></div>\",\"PeriodicalId\":10650,\"journal\":{\"name\":\"Computational Materials Science\",\"volume\":\"244 \",\"pages\":\"Article 113267\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2024-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Computational Materials Science\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0927025624004889\",\"RegionNum\":3,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/7/28 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"MATERIALS SCIENCE, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational Materials Science","FirstCategoryId":"88","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0927025624004889","RegionNum":3,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/28 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
摘要
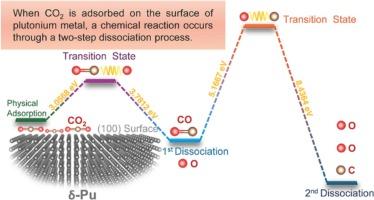
这项研究利用最先进的第一原理计算,阐明了一氧化碳和一氧化碳分子与 δ-Pu (100) 表面的吸附和解离相互作用。研究发现,空心位点是一氧化碳和一氧化碳最有利的吸附位置。在最重要的吸附构型中,两种物质都倾向于进行解离吸附,其特征是一氧化碳裂解成一个 O 原子和一个伴随的 CO 分子,而一氧化碳则分解成离散的 C 原子和 O 原子。根据计算,有利于这种构型的解离能垒分别为 CO 3.0568 eV 和 CO 5.1667 eV。对电子电荷和状态密度的全面分析表明,在解离过程中,电子从钚表面原子转移到碳质吸附物上。C-O 键解离后,钚的 6 个轨道与碳的 2 个轨道发生电子杂化,最终形成离子键。
Reaction mechanism of CO2 on the surface δ-Pu(1 0 0):a DFT study
This investigation elucidates the adsorptive and dissociative interactions of CO2 and CO molecules with the δ-Pu (1 0 0) surface, utilizing state-of-the-art first-principles calculations. The study unearths that the hollow site epitomizes the most favorable adsorption locus for both CO2 and CO. Within the paramount adsorption configuration, there prevails a propensity for both species to partake in dissociative adsorption, characterized by CO2 cleaving into an O atom and a concomitant CO moiety, whereas CO disintegrates into discrete C and O atoms. The dissociation energy barriers conducive to such configurations are computed to be 3.0568 eV for CO2 and 5.1667 eV for CO. A thorough analysis of electronic charges and density of states intimates a transfer of electrons from the Plutonium surface atoms to the carbonaceous adsorbates during dissociation. Post dissociation of the C-O bond, the 6d orbitals of Plutonium engage in electronic hybridization with the 2p orbitals of Carbon, culminating in the constitution of ionic bonding.
期刊介绍:
The goal of Computational Materials Science is to report on results that provide new or unique insights into, or significantly expand our understanding of, the properties of materials or phenomena associated with their design, synthesis, processing, characterization, and utilization. To be relevant to the journal, the results should be applied or applicable to specific material systems that are discussed within the submission.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
