Ernest Oduro-Kwateng, Musab Ali, Ibrahim Oluwatobi Kehinde, Zhichao Zhang, Mahmoud E. S. Soliman
{"title":"通过绘制 PP 界面的相互作用基元和物理化学过滤,从新合理设计基于肽的蛋白质-蛋白质抑制剂(Pep-PPIs)方法:以 p25-Cdk5 介导的神经退行性疾病为例。","authors":"Ernest Oduro-Kwateng, Musab Ali, Ibrahim Oluwatobi Kehinde, Zhichao Zhang, Mahmoud E. S. Soliman","doi":"10.1002/jcb.30633","DOIUrl":null,"url":null,"abstract":"<p>Protein–protein interactions, or PPIs, are a part of every biological activity and have been linked to a number of diseases, including cancer, infectious diseases, and neurological disorders. As such, targeting PPIs is considered a strategic and vital approach in the development of new medications. Nonetheless, the wide and flat contact interface makes it difficult to find small-molecule PP inhibitors. An alternative strategy would be to use the PPI interaction motifs as building blocks for the design of peptide-based inhibitors. Herein, we designed 12-mer peptide inhibitors to target p25-inducing-cyclin-dependent kinase (Cdk5) hyperregulation, a PPI that has been shown to perpetuate neuroinflammation, which is one of the major causal implications of neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, and frontotemporal dementia. We generated a library of 5 062 500 peptide combination sequences (PCS) derived from the interaction motif of Cdk5/p25 PP interface. The 20 amino acids were differentiated into six groups, namely, hydrophobic (aliphatic), aromatic, basic, acidic, unique, and polar uncharged, on the basis of their physiochemical properties. To preserve the interaction motif necessary for ideal binding, de novo modeling of all possible peptide sequence substitutions was considered. A set of filters, backed by the Support Vector Machine (SVM) algorithm, was then used to create a shortlisted custom peptide library that met specific bioavailability, toxicity, and therapeutic relevance, leading to a refined library of 15 PCS. A greedy algorithm and coarse-grained force field were used to predict peptide structure and folding before subsequent modeling studies. Molecular docking was performed to estimate the relative binding affinities, and out of the top hits, Pep15 was subjected to molecular dynamics simulations and binding free-energy calculations in comparison to a known peptide inhibitor with experimental data (template peptide). Interestingly, the identified peptide through our protocol, Pep15, was found to show a significantly higher binding affinity than the reference template peptide (−48.10 ± 0.23 kcal/mol and −17.53 ± 0.27 kcal/mol, respectively). In comparison to the template peptide, Pep15 was found to possess a more compact and buried surface area, tighter binding landscape, and reduced conformational variability, leading to enhanced structural and kinetic stability of the Cdk5/p25 complex. Notably, both peptide inhibitors were found to have a minimal impact on the architectural integrity of the Cdk5/p25 secondary structure.</p><p>Herein, we propose Pep15 as a novel and potentially disruptive peptide drug for Cdk5/p25-mediated neurodegenerative phenotypes that require further clinical investigation. The systematic protocol and findings of this report would serve as a valuable tool in the identification of critical PPI interface reactive residues, designing of analogs, and identification of more potent peptide-based PPI inhibitors.</p>","PeriodicalId":15219,"journal":{"name":"Journal of cellular biochemistry","volume":"125 9","pages":""},"PeriodicalIF":3.0000,"publicationDate":"2024-08-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcb.30633","citationCount":"0","resultStr":"{\"title\":\"De Novo Rational Design of Peptide-Based Protein–Protein Inhibitors (Pep-PPIs) Approach by Mapping the Interaction Motifs of the PP Interface and Physicochemical Filtration: A Case on p25-Cdk5-Mediated Neurodegenerative Diseases\",\"authors\":\"Ernest Oduro-Kwateng, Musab Ali, Ibrahim Oluwatobi Kehinde, Zhichao Zhang, Mahmoud E. S. Soliman\",\"doi\":\"10.1002/jcb.30633\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Protein–protein interactions, or PPIs, are a part of every biological activity and have been linked to a number of diseases, including cancer, infectious diseases, and neurological disorders. As such, targeting PPIs is considered a strategic and vital approach in the development of new medications. Nonetheless, the wide and flat contact interface makes it difficult to find small-molecule PP inhibitors. An alternative strategy would be to use the PPI interaction motifs as building blocks for the design of peptide-based inhibitors. Herein, we designed 12-mer peptide inhibitors to target p25-inducing-cyclin-dependent kinase (Cdk5) hyperregulation, a PPI that has been shown to perpetuate neuroinflammation, which is one of the major causal implications of neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, and frontotemporal dementia. We generated a library of 5 062 500 peptide combination sequences (PCS) derived from the interaction motif of Cdk5/p25 PP interface. The 20 amino acids were differentiated into six groups, namely, hydrophobic (aliphatic), aromatic, basic, acidic, unique, and polar uncharged, on the basis of their physiochemical properties. To preserve the interaction motif necessary for ideal binding, de novo modeling of all possible peptide sequence substitutions was considered. A set of filters, backed by the Support Vector Machine (SVM) algorithm, was then used to create a shortlisted custom peptide library that met specific bioavailability, toxicity, and therapeutic relevance, leading to a refined library of 15 PCS. A greedy algorithm and coarse-grained force field were used to predict peptide structure and folding before subsequent modeling studies. Molecular docking was performed to estimate the relative binding affinities, and out of the top hits, Pep15 was subjected to molecular dynamics simulations and binding free-energy calculations in comparison to a known peptide inhibitor with experimental data (template peptide). Interestingly, the identified peptide through our protocol, Pep15, was found to show a significantly higher binding affinity than the reference template peptide (−48.10 ± 0.23 kcal/mol and −17.53 ± 0.27 kcal/mol, respectively). In comparison to the template peptide, Pep15 was found to possess a more compact and buried surface area, tighter binding landscape, and reduced conformational variability, leading to enhanced structural and kinetic stability of the Cdk5/p25 complex. Notably, both peptide inhibitors were found to have a minimal impact on the architectural integrity of the Cdk5/p25 secondary structure.</p><p>Herein, we propose Pep15 as a novel and potentially disruptive peptide drug for Cdk5/p25-mediated neurodegenerative phenotypes that require further clinical investigation. The systematic protocol and findings of this report would serve as a valuable tool in the identification of critical PPI interface reactive residues, designing of analogs, and identification of more potent peptide-based PPI inhibitors.</p>\",\"PeriodicalId\":15219,\"journal\":{\"name\":\"Journal of cellular biochemistry\",\"volume\":\"125 9\",\"pages\":\"\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-08-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcb.30633\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of cellular biochemistry\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcb.30633\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of cellular biochemistry","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcb.30633","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
De Novo Rational Design of Peptide-Based Protein–Protein Inhibitors (Pep-PPIs) Approach by Mapping the Interaction Motifs of the PP Interface and Physicochemical Filtration: A Case on p25-Cdk5-Mediated Neurodegenerative Diseases
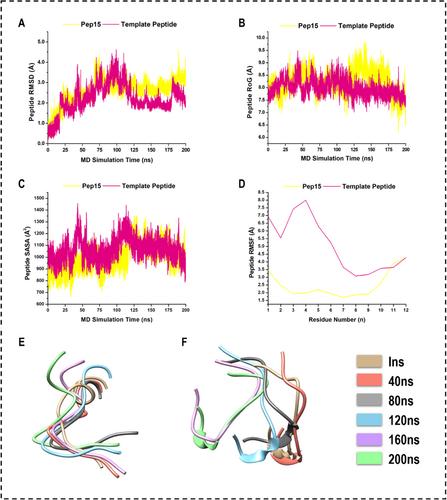
Protein–protein interactions, or PPIs, are a part of every biological activity and have been linked to a number of diseases, including cancer, infectious diseases, and neurological disorders. As such, targeting PPIs is considered a strategic and vital approach in the development of new medications. Nonetheless, the wide and flat contact interface makes it difficult to find small-molecule PP inhibitors. An alternative strategy would be to use the PPI interaction motifs as building blocks for the design of peptide-based inhibitors. Herein, we designed 12-mer peptide inhibitors to target p25-inducing-cyclin-dependent kinase (Cdk5) hyperregulation, a PPI that has been shown to perpetuate neuroinflammation, which is one of the major causal implications of neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, and frontotemporal dementia. We generated a library of 5 062 500 peptide combination sequences (PCS) derived from the interaction motif of Cdk5/p25 PP interface. The 20 amino acids were differentiated into six groups, namely, hydrophobic (aliphatic), aromatic, basic, acidic, unique, and polar uncharged, on the basis of their physiochemical properties. To preserve the interaction motif necessary for ideal binding, de novo modeling of all possible peptide sequence substitutions was considered. A set of filters, backed by the Support Vector Machine (SVM) algorithm, was then used to create a shortlisted custom peptide library that met specific bioavailability, toxicity, and therapeutic relevance, leading to a refined library of 15 PCS. A greedy algorithm and coarse-grained force field were used to predict peptide structure and folding before subsequent modeling studies. Molecular docking was performed to estimate the relative binding affinities, and out of the top hits, Pep15 was subjected to molecular dynamics simulations and binding free-energy calculations in comparison to a known peptide inhibitor with experimental data (template peptide). Interestingly, the identified peptide through our protocol, Pep15, was found to show a significantly higher binding affinity than the reference template peptide (−48.10 ± 0.23 kcal/mol and −17.53 ± 0.27 kcal/mol, respectively). In comparison to the template peptide, Pep15 was found to possess a more compact and buried surface area, tighter binding landscape, and reduced conformational variability, leading to enhanced structural and kinetic stability of the Cdk5/p25 complex. Notably, both peptide inhibitors were found to have a minimal impact on the architectural integrity of the Cdk5/p25 secondary structure.
Herein, we propose Pep15 as a novel and potentially disruptive peptide drug for Cdk5/p25-mediated neurodegenerative phenotypes that require further clinical investigation. The systematic protocol and findings of this report would serve as a valuable tool in the identification of critical PPI interface reactive residues, designing of analogs, and identification of more potent peptide-based PPI inhibitors.
期刊介绍:
The Journal of Cellular Biochemistry publishes descriptions of original research in which complex cellular, pathogenic, clinical, or animal model systems are studied by biochemical, molecular, genetic, epigenetic or quantitative ultrastructural approaches. Submission of papers reporting genomic, proteomic, bioinformatics and systems biology approaches to identify and characterize parameters of biological control in a cellular context are encouraged. The areas covered include, but are not restricted to, conditions, agents, regulatory networks, or differentiation states that influence structure, cell cycle & growth control, structure-function relationships.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
