Yifan Zhang , Rajni Chahal , M. Mustafa Azeem , Stephen Lam , Karl Ludwig , Uday Pal , Michael C. Gao , Adam Powell , Yu Zhong
{"title":"对 CaF2-MgF2 二元氟化物体系在高温下的结构、热力学和传输特性的计算见解","authors":"Yifan Zhang , Rajni Chahal , M. Mustafa Azeem , Stephen Lam , Karl Ludwig , Uday Pal , Michael C. Gao , Adam Powell , Yu Zhong","doi":"10.1016/j.commatsci.2024.113294","DOIUrl":null,"url":null,"abstract":"<div><p>The structural, thermodynamic and transport properties of the CaF<sub>2</sub>-MgF<sub>2</sub> molten salt system were investigated with ab initio molecular dynamics (AIMD), system-specific neural network interatomic potentials (NNIPs) and universal PreFerred Potentials (PFP). We trained an NNIP model using AIMD data as input and used this potential to efficiently simulate the interactions within a large supercell in a temperature range of 1273–1773 K. The Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) code was employed to validate our trained NNIP model. The Matlantis software with universal PFP is also presented to prove its feasibility for MD calculations and can be considered as a useful alternative simulation tool for higher-order systems where existing potentials are not readily available. We calculated structural and thermodynamic properties including radial distribution function (RDF), angular distribution function (ADF), specific heat capacity, ionic self-diffusivity, and viscosity. Our results indicate that the system exhibited a high degree of structural disorder, with the Ca, Mg, and F ions forming a liquid solution. Using PFP, the positions of the first peak in RDFs for Ca-F and Mg-F pairs are only slightly left-shifted (<0.05 Å), and the estimated viscosity of the melt decreases from 4.613 mPa·s to 1.846 mPa·s with an increase in temperature from 1273 K to 1773 K, in agreement with the NNIP trained specifically for CaF<sub>2</sub>-MgF<sub>2</sub>. Our results provide valuable insights into the properties of the CaF<sub>2</sub>-MgF<sub>2</sub> system at high temperatures and serve as predictive models for the development of new electrolytes that could be used for silicon epitaxy by adding silica.</p></div>","PeriodicalId":10650,"journal":{"name":"Computational Materials Science","volume":"245 ","pages":"Article 113294"},"PeriodicalIF":3.3000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Computational insights into the structural, thermodynamic and transport properties of CaF2-MgF2 binary fluoride system at high temperatures\",\"authors\":\"Yifan Zhang , Rajni Chahal , M. Mustafa Azeem , Stephen Lam , Karl Ludwig , Uday Pal , Michael C. Gao , Adam Powell , Yu Zhong\",\"doi\":\"10.1016/j.commatsci.2024.113294\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The structural, thermodynamic and transport properties of the CaF<sub>2</sub>-MgF<sub>2</sub> molten salt system were investigated with ab initio molecular dynamics (AIMD), system-specific neural network interatomic potentials (NNIPs) and universal PreFerred Potentials (PFP). We trained an NNIP model using AIMD data as input and used this potential to efficiently simulate the interactions within a large supercell in a temperature range of 1273–1773 K. The Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) code was employed to validate our trained NNIP model. The Matlantis software with universal PFP is also presented to prove its feasibility for MD calculations and can be considered as a useful alternative simulation tool for higher-order systems where existing potentials are not readily available. We calculated structural and thermodynamic properties including radial distribution function (RDF), angular distribution function (ADF), specific heat capacity, ionic self-diffusivity, and viscosity. Our results indicate that the system exhibited a high degree of structural disorder, with the Ca, Mg, and F ions forming a liquid solution. Using PFP, the positions of the first peak in RDFs for Ca-F and Mg-F pairs are only slightly left-shifted (<0.05 Å), and the estimated viscosity of the melt decreases from 4.613 mPa·s to 1.846 mPa·s with an increase in temperature from 1273 K to 1773 K, in agreement with the NNIP trained specifically for CaF<sub>2</sub>-MgF<sub>2</sub>. Our results provide valuable insights into the properties of the CaF<sub>2</sub>-MgF<sub>2</sub> system at high temperatures and serve as predictive models for the development of new electrolytes that could be used for silicon epitaxy by adding silica.</p></div>\",\"PeriodicalId\":10650,\"journal\":{\"name\":\"Computational Materials Science\",\"volume\":\"245 \",\"pages\":\"Article 113294\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2024-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Computational Materials Science\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0927025624005159\",\"RegionNum\":3,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/8/24 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"MATERIALS SCIENCE, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational Materials Science","FirstCategoryId":"88","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0927025624005159","RegionNum":3,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/24 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
Computational insights into the structural, thermodynamic and transport properties of CaF2-MgF2 binary fluoride system at high temperatures
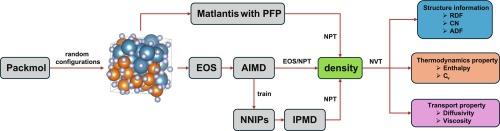
The structural, thermodynamic and transport properties of the CaF2-MgF2 molten salt system were investigated with ab initio molecular dynamics (AIMD), system-specific neural network interatomic potentials (NNIPs) and universal PreFerred Potentials (PFP). We trained an NNIP model using AIMD data as input and used this potential to efficiently simulate the interactions within a large supercell in a temperature range of 1273–1773 K. The Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) code was employed to validate our trained NNIP model. The Matlantis software with universal PFP is also presented to prove its feasibility for MD calculations and can be considered as a useful alternative simulation tool for higher-order systems where existing potentials are not readily available. We calculated structural and thermodynamic properties including radial distribution function (RDF), angular distribution function (ADF), specific heat capacity, ionic self-diffusivity, and viscosity. Our results indicate that the system exhibited a high degree of structural disorder, with the Ca, Mg, and F ions forming a liquid solution. Using PFP, the positions of the first peak in RDFs for Ca-F and Mg-F pairs are only slightly left-shifted (<0.05 Å), and the estimated viscosity of the melt decreases from 4.613 mPa·s to 1.846 mPa·s with an increase in temperature from 1273 K to 1773 K, in agreement with the NNIP trained specifically for CaF2-MgF2. Our results provide valuable insights into the properties of the CaF2-MgF2 system at high temperatures and serve as predictive models for the development of new electrolytes that could be used for silicon epitaxy by adding silica.
期刊介绍:
The goal of Computational Materials Science is to report on results that provide new or unique insights into, or significantly expand our understanding of, the properties of materials or phenomena associated with their design, synthesis, processing, characterization, and utilization. To be relevant to the journal, the results should be applied or applicable to specific material systems that are discussed within the submission.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
