Jun Luo, Omar Ben Said, Peigen Xie, Marco Gibaldi, Jake Burner, Cécile Pereira, Tom K. Woo
{"title":"MEPO-ML:用于快速生成金属有机框架中部分原子电荷的稳健图注意网络模型","authors":"Jun Luo, Omar Ben Said, Peigen Xie, Marco Gibaldi, Jake Burner, Cécile Pereira, Tom K. Woo","doi":"10.1038/s41524-024-01413-4","DOIUrl":null,"url":null,"abstract":"<p>Accurate computation of the gas adsorption properties of MOFs is usually bottlenecked by the DFT calculations required to generate partial atomic charges. Therefore, large virtual screenings of MOFs often use the QEq method which is rapid, but of limited accuracy. Recently, machine learning (ML) models have been trained to generate charges in much better agreement with DFT-derived charges compared to the QEq models. Previous ML charge models for MOFs have all used training sets with less than 3000 MOFs obtained from the CoRE MOF database, which has recently been shown to have high structural error rates. In this work, we developed a graph attention network model for predicting DFT-derived charges in MOFs where the model was developed with the ARC-MOF database that contains 279,632 MOFs and over 40 million charges. This model, which we call <i>MEPO-ML</i>, predicts charges with a mean absolute error of 0.025e on our test set of over 27 K MOFs. Other ML models reported in the literature were also trained using the same dataset and descriptors, and MEPO-ML was shown to give the lowest errors. The gas adsorption properties evaluated using MEPO-ML charges are found to be in significantly better agreement with the reference DFT-derived charges compared to the empirical charges, for both polar and non-polar gases. Using only a single CPU core on our benchmark computer, MEPO-ML charges can be generated in less than two seconds on average (including all computations required to apply the model) for MOFs in the test set of 27 K MOFs.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"7 1","pages":""},"PeriodicalIF":9.4000,"publicationDate":"2024-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"MEPO-ML: a robust graph attention network model for rapid generation of partial atomic charges in metal-organic frameworks\",\"authors\":\"Jun Luo, Omar Ben Said, Peigen Xie, Marco Gibaldi, Jake Burner, Cécile Pereira, Tom K. Woo\",\"doi\":\"10.1038/s41524-024-01413-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Accurate computation of the gas adsorption properties of MOFs is usually bottlenecked by the DFT calculations required to generate partial atomic charges. Therefore, large virtual screenings of MOFs often use the QEq method which is rapid, but of limited accuracy. Recently, machine learning (ML) models have been trained to generate charges in much better agreement with DFT-derived charges compared to the QEq models. Previous ML charge models for MOFs have all used training sets with less than 3000 MOFs obtained from the CoRE MOF database, which has recently been shown to have high structural error rates. In this work, we developed a graph attention network model for predicting DFT-derived charges in MOFs where the model was developed with the ARC-MOF database that contains 279,632 MOFs and over 40 million charges. This model, which we call <i>MEPO-ML</i>, predicts charges with a mean absolute error of 0.025e on our test set of over 27 K MOFs. Other ML models reported in the literature were also trained using the same dataset and descriptors, and MEPO-ML was shown to give the lowest errors. The gas adsorption properties evaluated using MEPO-ML charges are found to be in significantly better agreement with the reference DFT-derived charges compared to the empirical charges, for both polar and non-polar gases. Using only a single CPU core on our benchmark computer, MEPO-ML charges can be generated in less than two seconds on average (including all computations required to apply the model) for MOFs in the test set of 27 K MOFs.</p>\",\"PeriodicalId\":19342,\"journal\":{\"name\":\"npj Computational Materials\",\"volume\":\"7 1\",\"pages\":\"\"},\"PeriodicalIF\":9.4000,\"publicationDate\":\"2024-09-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Computational Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1038/s41524-024-01413-4\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01413-4","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
MEPO-ML: a robust graph attention network model for rapid generation of partial atomic charges in metal-organic frameworks
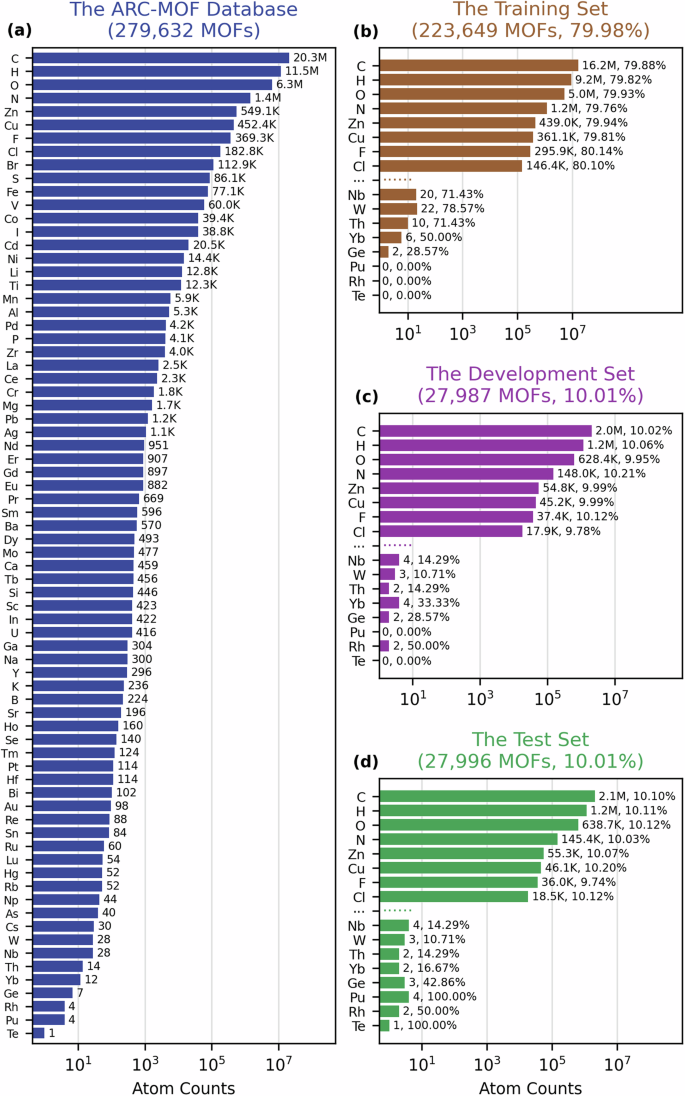
Accurate computation of the gas adsorption properties of MOFs is usually bottlenecked by the DFT calculations required to generate partial atomic charges. Therefore, large virtual screenings of MOFs often use the QEq method which is rapid, but of limited accuracy. Recently, machine learning (ML) models have been trained to generate charges in much better agreement with DFT-derived charges compared to the QEq models. Previous ML charge models for MOFs have all used training sets with less than 3000 MOFs obtained from the CoRE MOF database, which has recently been shown to have high structural error rates. In this work, we developed a graph attention network model for predicting DFT-derived charges in MOFs where the model was developed with the ARC-MOF database that contains 279,632 MOFs and over 40 million charges. This model, which we call MEPO-ML, predicts charges with a mean absolute error of 0.025e on our test set of over 27 K MOFs. Other ML models reported in the literature were also trained using the same dataset and descriptors, and MEPO-ML was shown to give the lowest errors. The gas adsorption properties evaluated using MEPO-ML charges are found to be in significantly better agreement with the reference DFT-derived charges compared to the empirical charges, for both polar and non-polar gases. Using only a single CPU core on our benchmark computer, MEPO-ML charges can be generated in less than two seconds on average (including all computations required to apply the model) for MOFs in the test set of 27 K MOFs.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
