{"title":"iBhb-Lys:使用自动编码器特征表示和模糊 SVM 算法识别赖氨酸 β-羟基丁酰化位点。","authors":"Zhe Ju, Qing-Bao Zhang","doi":"10.1016/j.ab.2024.115715","DOIUrl":null,"url":null,"abstract":"<div><div>Lysine β-hydroxybutyrylation (Kbhb) is newly discovered β-hydroxybutyrylate-derived histone modification which has been associated with the pathogenesis of many human diseases. To further elucidate the biological significance and molecular mechanism of Kbhb, it is necessary to accurately identify the Kbhb sites from protein sequences. In this study, a novel computational model named iBhb-Lys is developed for the identification of Kbhb sites. Four types of features are combined to encode each Kbhb site as a 3266-dimensional feature vector. And the autoencoder network is used to reduce the dimensionality of feature space, due to the high dimensionality of the combined features. In addition, to effectively reduce the influence of noise and outlier on classification, a new fuzzy support vector machine algorithm is proposed by incorporating the density around the sample into the fuzzy membership function. As illustrated by independent test, the AUC value of iBhb-Lys has increased by 2.22 % compared to the existing predictor KbhbXG. Feature analysis shows that some amino acid composition features, such as the occurrence frequency of leucine and histidine residues around Kbhb sites, contribute profoundly to the identification of Kbhb sites. The conclusions drawn in this study may provide useful reference for studying the molecular mechanism of Kbhb.</div></div>","PeriodicalId":7830,"journal":{"name":"Analytical biochemistry","volume":"697 ","pages":"Article 115715"},"PeriodicalIF":2.5000,"publicationDate":"2025-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"iBhb-Lys: Identify lysine β-hydroxybutyrylation sites using autoencoder feature representation and fuzzy SVM algorithm\",\"authors\":\"Zhe Ju, Qing-Bao Zhang\",\"doi\":\"10.1016/j.ab.2024.115715\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Lysine β-hydroxybutyrylation (Kbhb) is newly discovered β-hydroxybutyrylate-derived histone modification which has been associated with the pathogenesis of many human diseases. To further elucidate the biological significance and molecular mechanism of Kbhb, it is necessary to accurately identify the Kbhb sites from protein sequences. In this study, a novel computational model named iBhb-Lys is developed for the identification of Kbhb sites. Four types of features are combined to encode each Kbhb site as a 3266-dimensional feature vector. And the autoencoder network is used to reduce the dimensionality of feature space, due to the high dimensionality of the combined features. In addition, to effectively reduce the influence of noise and outlier on classification, a new fuzzy support vector machine algorithm is proposed by incorporating the density around the sample into the fuzzy membership function. As illustrated by independent test, the AUC value of iBhb-Lys has increased by 2.22 % compared to the existing predictor KbhbXG. Feature analysis shows that some amino acid composition features, such as the occurrence frequency of leucine and histidine residues around Kbhb sites, contribute profoundly to the identification of Kbhb sites. The conclusions drawn in this study may provide useful reference for studying the molecular mechanism of Kbhb.</div></div>\",\"PeriodicalId\":7830,\"journal\":{\"name\":\"Analytical biochemistry\",\"volume\":\"697 \",\"pages\":\"Article 115715\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2025-02-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Analytical biochemistry\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0003269724002598\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/11/7 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Analytical biochemistry","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0003269724002598","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/7 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
iBhb-Lys: Identify lysine β-hydroxybutyrylation sites using autoencoder feature representation and fuzzy SVM algorithm
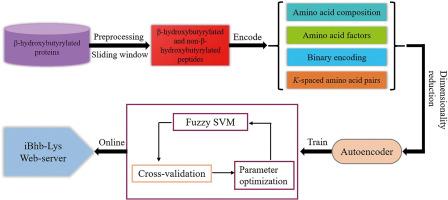
Lysine β-hydroxybutyrylation (Kbhb) is newly discovered β-hydroxybutyrylate-derived histone modification which has been associated with the pathogenesis of many human diseases. To further elucidate the biological significance and molecular mechanism of Kbhb, it is necessary to accurately identify the Kbhb sites from protein sequences. In this study, a novel computational model named iBhb-Lys is developed for the identification of Kbhb sites. Four types of features are combined to encode each Kbhb site as a 3266-dimensional feature vector. And the autoencoder network is used to reduce the dimensionality of feature space, due to the high dimensionality of the combined features. In addition, to effectively reduce the influence of noise and outlier on classification, a new fuzzy support vector machine algorithm is proposed by incorporating the density around the sample into the fuzzy membership function. As illustrated by independent test, the AUC value of iBhb-Lys has increased by 2.22 % compared to the existing predictor KbhbXG. Feature analysis shows that some amino acid composition features, such as the occurrence frequency of leucine and histidine residues around Kbhb sites, contribute profoundly to the identification of Kbhb sites. The conclusions drawn in this study may provide useful reference for studying the molecular mechanism of Kbhb.
期刊介绍:
The journal''s title Analytical Biochemistry: Methods in the Biological Sciences declares its broad scope: methods for the basic biological sciences that include biochemistry, molecular genetics, cell biology, proteomics, immunology, bioinformatics and wherever the frontiers of research take the field.
The emphasis is on methods from the strictly analytical to the more preparative that would include novel approaches to protein purification as well as improvements in cell and organ culture. The actual techniques are equally inclusive ranging from aptamers to zymology.
The journal has been particularly active in:
-Analytical techniques for biological molecules-
Aptamer selection and utilization-
Biosensors-
Chromatography-
Cloning, sequencing and mutagenesis-
Electrochemical methods-
Electrophoresis-
Enzyme characterization methods-
Immunological approaches-
Mass spectrometry of proteins and nucleic acids-
Metabolomics-
Nano level techniques-
Optical spectroscopy in all its forms.
The journal is reluctant to include most drug and strictly clinical studies as there are more suitable publication platforms for these types of papers.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
