Jin-Hee Seo, Jang-Gyu Choi, Hyun-Jin Park, Ji-Hong Cho, Young-Eun Park, Ju-Sung Im, Su-Young Hong, Kwang-Soo Cho
{"title":"韩国疫霉菌分离物线粒体全基因组序列及线粒体单倍型比较分析。","authors":"Jin-Hee Seo, Jang-Gyu Choi, Hyun-Jin Park, Ji-Hong Cho, Young-Eun Park, Ju-Sung Im, Su-Young Hong, Kwang-Soo Cho","doi":"10.5423/PPJ.OA.07.2022.0093","DOIUrl":null,"url":null,"abstract":"<p><p>Potato late blight caused by Phytophthora infestans is a destructive disease in Korea. To elucidate the genomic variation of the mitochondrial (mt) genome, we assembled its complete mt genome and compared its sequence among different haplotypes. The mt genome sequences of four Korean P. infestans isolates were revealed by Illumina HiSeq. The size of the circular mt genome of the four major genotypes, KR_1_A1, KR_2_A2, SIB-1, and US-11, was 39,872, 39,836, 39,872, and 39,840 bp, respectively. All genotypes contained the same 61 genes in the same order, comprising two RNA-encoding genes, 16 ribosomal genes, 25 transfer RNA, 17 genes encoding electron transport and ATP synthesis, 11 open reading frames of unknown function, and one protein import-related gene, tatC. The coding region comprised 91% of the genome, and GC content was 22.3%. The haplotypes were further analyzed based on sequence polymorphism at two hypervariable regions (HVRi), carrying a 2 kb insertion/deletion sequence, and HVRii, carrying 36 bp variable number tandem repeats (VNTRs). All four genotypes carried the 2 kb insertion/deletion sequence in HVRi, whereas HVRii had two VNTRs in KR_1_A1 and SIB-1 but three VNTRs in US-11 and KR_2_A2. Minimal spanning network and phylogenetic analysis based on 5,814 bp of mtDNA sequences from five loci, KR_1_A1 and SIB-1 were classified as IIa-6 haplotype, and isolates KR_1_A2 and US-11 as haplotypes IIa-5 and IIb-2, respectively. mtDNA sequences of KR_1_A1 and SIB-1 shared 100% sequence identity, and both were 99.9% similar to those of KR_2_A2 and US-11.</p>","PeriodicalId":20173,"journal":{"name":"Plant Pathology Journal","volume":"38 5","pages":"541-549"},"PeriodicalIF":2.5000,"publicationDate":"2022-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/87/fb/ppj-oa-07-2022-0093.PMC9561156.pdf","citationCount":"0","resultStr":"{\"title\":\"Complete Mitochondrial Genome Sequences of Korean Phytophthora infestans Isolates and Comparative Analysis of Mitochondrial Haplotypes.\",\"authors\":\"Jin-Hee Seo, Jang-Gyu Choi, Hyun-Jin Park, Ji-Hong Cho, Young-Eun Park, Ju-Sung Im, Su-Young Hong, Kwang-Soo Cho\",\"doi\":\"10.5423/PPJ.OA.07.2022.0093\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Potato late blight caused by Phytophthora infestans is a destructive disease in Korea. To elucidate the genomic variation of the mitochondrial (mt) genome, we assembled its complete mt genome and compared its sequence among different haplotypes. The mt genome sequences of four Korean P. infestans isolates were revealed by Illumina HiSeq. The size of the circular mt genome of the four major genotypes, KR_1_A1, KR_2_A2, SIB-1, and US-11, was 39,872, 39,836, 39,872, and 39,840 bp, respectively. All genotypes contained the same 61 genes in the same order, comprising two RNA-encoding genes, 16 ribosomal genes, 25 transfer RNA, 17 genes encoding electron transport and ATP synthesis, 11 open reading frames of unknown function, and one protein import-related gene, tatC. The coding region comprised 91% of the genome, and GC content was 22.3%. The haplotypes were further analyzed based on sequence polymorphism at two hypervariable regions (HVRi), carrying a 2 kb insertion/deletion sequence, and HVRii, carrying 36 bp variable number tandem repeats (VNTRs). All four genotypes carried the 2 kb insertion/deletion sequence in HVRi, whereas HVRii had two VNTRs in KR_1_A1 and SIB-1 but three VNTRs in US-11 and KR_2_A2. Minimal spanning network and phylogenetic analysis based on 5,814 bp of mtDNA sequences from five loci, KR_1_A1 and SIB-1 were classified as IIa-6 haplotype, and isolates KR_1_A2 and US-11 as haplotypes IIa-5 and IIb-2, respectively. mtDNA sequences of KR_1_A1 and SIB-1 shared 100% sequence identity, and both were 99.9% similar to those of KR_2_A2 and US-11.</p>\",\"PeriodicalId\":20173,\"journal\":{\"name\":\"Plant Pathology Journal\",\"volume\":\"38 5\",\"pages\":\"541-549\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2022-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/87/fb/ppj-oa-07-2022-0093.PMC9561156.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Plant Pathology Journal\",\"FirstCategoryId\":\"97\",\"ListUrlMain\":\"https://doi.org/10.5423/PPJ.OA.07.2022.0093\",\"RegionNum\":3,\"RegionCategory\":\"农林科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"PLANT SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Plant Pathology Journal","FirstCategoryId":"97","ListUrlMain":"https://doi.org/10.5423/PPJ.OA.07.2022.0093","RegionNum":3,"RegionCategory":"农林科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"PLANT SCIENCES","Score":null,"Total":0}
引用次数: 0
摘要
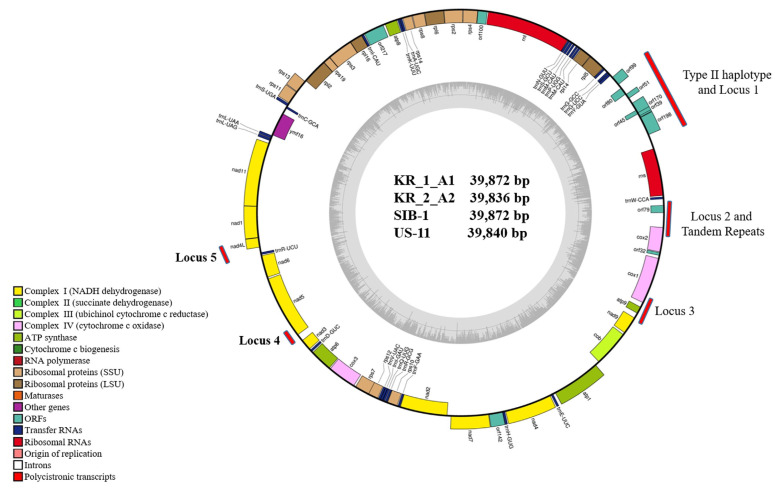
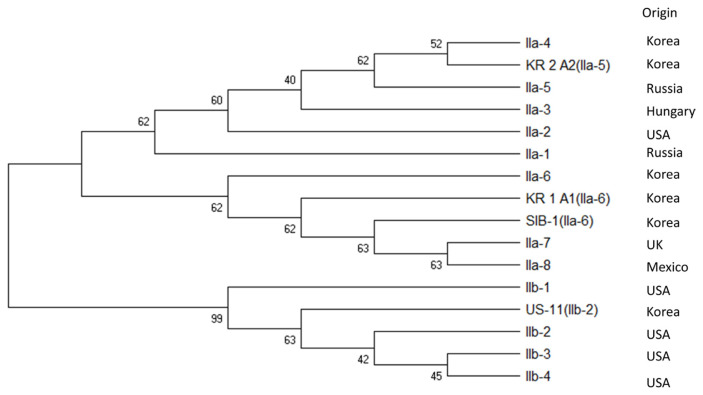
马铃薯晚疫病是由马铃薯疫霉引起的马铃薯疫病。为了阐明线粒体(mt)基因组的基因组变异,我们组装了其完整的mt基因组,并比较了其在不同单倍型中的序列。利用Illumina HiSeq软件分析了韩国4株病原菌的mt基因组序列。4个主要基因型KR_1_A1、KR_2_A2、sib1和US-11的环mt基因组大小分别为39,872、39,836、39,872和39,840 bp。所有基因型均包含相同的61个基因,顺序相同,包括2个RNA编码基因,16个核糖体基因,25个转移RNA, 17个电子传递和ATP合成基因,11个功能未知的开放阅读框,1个蛋白质输入相关基因tatC。编码区占基因组的91%,GC含量为22.3%。单倍型在两个携带2kb插入/删除序列的高变区(HVRi)和携带36bp可变数串联重复序列(VNTRs)的HVRii进行序列多态性分析。所有4种基因型在HVRi中都携带2kb的插入/缺失序列,而HVRii在KR_1_A1和sib1中有2个VNTRs,在US-11和KR_2_A2中有3个VNTRs。基于5个位点的5814 bp mtDNA序列的最小跨越网络和系统发育分析,将KR_1_A1和ib -1分类为IIa-6单倍型,分离出KR_1_A2和US-11分别为IIa-5和IIb-2单倍型。KR_1_A1和sb -1的mtDNA序列同源性为100%,与KR_2_A2和US-11的相似度为99.9%。
Complete Mitochondrial Genome Sequences of Korean Phytophthora infestans Isolates and Comparative Analysis of Mitochondrial Haplotypes.
Potato late blight caused by Phytophthora infestans is a destructive disease in Korea. To elucidate the genomic variation of the mitochondrial (mt) genome, we assembled its complete mt genome and compared its sequence among different haplotypes. The mt genome sequences of four Korean P. infestans isolates were revealed by Illumina HiSeq. The size of the circular mt genome of the four major genotypes, KR_1_A1, KR_2_A2, SIB-1, and US-11, was 39,872, 39,836, 39,872, and 39,840 bp, respectively. All genotypes contained the same 61 genes in the same order, comprising two RNA-encoding genes, 16 ribosomal genes, 25 transfer RNA, 17 genes encoding electron transport and ATP synthesis, 11 open reading frames of unknown function, and one protein import-related gene, tatC. The coding region comprised 91% of the genome, and GC content was 22.3%. The haplotypes were further analyzed based on sequence polymorphism at two hypervariable regions (HVRi), carrying a 2 kb insertion/deletion sequence, and HVRii, carrying 36 bp variable number tandem repeats (VNTRs). All four genotypes carried the 2 kb insertion/deletion sequence in HVRi, whereas HVRii had two VNTRs in KR_1_A1 and SIB-1 but three VNTRs in US-11 and KR_2_A2. Minimal spanning network and phylogenetic analysis based on 5,814 bp of mtDNA sequences from five loci, KR_1_A1 and SIB-1 were classified as IIa-6 haplotype, and isolates KR_1_A2 and US-11 as haplotypes IIa-5 and IIb-2, respectively. mtDNA sequences of KR_1_A1 and SIB-1 shared 100% sequence identity, and both were 99.9% similar to those of KR_2_A2 and US-11.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
