{"title":"基于单细胞转录组分析的结肠癌七基因预后标志物的鉴定和验证","authors":"Yang Zhou, Yang Guo, Yuanhe Wang","doi":"10.1049/syb2.12041","DOIUrl":null,"url":null,"abstract":"<p>Colon cancer (CC) is one of the most commonly diagnosed tumours worldwide. Single-cell RNA sequencing (scRNA-seq) can accurately reflect the heterogeneity within and between tumour cells and identify important genes associated with cancer development and growth. In this study, scRNA-seq was used to identify reliable prognostic biomarkers in CC. ScRNA-seq data of CC before and after 5-fluorouracil treatment were first downloaded from the Gene Expression Omnibus database. The data were pre-processed, and dimensionality reduction was performed using principal component analysis and t-distributed stochastic neighbour embedding algorithms. Additionally, the transcriptome data, somatic variant data, and clinical reports of patients with CC were obtained from The Cancer Genome Atlas database. Seven key genes were identified using Cox regression analysis and the least absolute shrinkage and selection operator method to establish signatures associated with CC prognoses. The identified signatures were validated on independent datasets, and somatic mutations and potential oncogenic pathways were further explored. Based on these features, gene signatures, and other clinical variables, a more effective predictive model nomogram for patients with CC was constructed, and a decision curve analysis was performed to assess the utility of the nomogram. A prognostic signature consisting of seven prognostic-related genes, including <i>CAV2</i>, <i>EREG</i>, <i>NGFRAP1</i>, <i>WBSCR22</i>, <i>SPINT2</i>, <i>CCDC28A</i>, and <i>BCL10</i>, was constructed and validated. The proficiency and credibility of the signature were verified in both internal and external datasets, and the results showed that the seven-gene signature could effectively predict the prognosis of patients with CC under various clinical conditions. A nomogram was then constructed based on features such as the RiskScore, patients' age, neoplasm stage, and tumor (T), nodes (N), and metastases (M) classification, and the nomogram had good clinical utility. Higher RiskScores were associated with a higher tumour mutational burden, which was confirmed to be a prognostic risk factor. Gene set enrichment analysis showed that high-score groups were enriched in ‘cytoplasmic DNA sensing’, ‘Extracellular matrix receptor interactions’, and ‘focal adhesion’, and low-score groups were enriched in ‘natural killer cell-mediated cytotoxicity’, and ‘T-cell receptor signalling pathways’, among other pathways. A robust seven-gene marker for CC was identified based on scRNA-seq data and was validated in multiple independent cohort studies. These findings provide a new potential marker to predict the prognosis of patients with CC.</p>","PeriodicalId":50379,"journal":{"name":"IET Systems Biology","volume":"16 2","pages":"72-83"},"PeriodicalIF":1.9000,"publicationDate":"2022-03-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ietresearch.onlinelibrary.wiley.com/doi/epdf/10.1049/syb2.12041","citationCount":"3","resultStr":"{\"title\":\"Identification and validation of a seven-gene prognostic marker in colon cancer based on single-cell transcriptome analysis\",\"authors\":\"Yang Zhou, Yang Guo, Yuanhe Wang\",\"doi\":\"10.1049/syb2.12041\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Colon cancer (CC) is one of the most commonly diagnosed tumours worldwide. Single-cell RNA sequencing (scRNA-seq) can accurately reflect the heterogeneity within and between tumour cells and identify important genes associated with cancer development and growth. In this study, scRNA-seq was used to identify reliable prognostic biomarkers in CC. ScRNA-seq data of CC before and after 5-fluorouracil treatment were first downloaded from the Gene Expression Omnibus database. The data were pre-processed, and dimensionality reduction was performed using principal component analysis and t-distributed stochastic neighbour embedding algorithms. Additionally, the transcriptome data, somatic variant data, and clinical reports of patients with CC were obtained from The Cancer Genome Atlas database. Seven key genes were identified using Cox regression analysis and the least absolute shrinkage and selection operator method to establish signatures associated with CC prognoses. The identified signatures were validated on independent datasets, and somatic mutations and potential oncogenic pathways were further explored. Based on these features, gene signatures, and other clinical variables, a more effective predictive model nomogram for patients with CC was constructed, and a decision curve analysis was performed to assess the utility of the nomogram. A prognostic signature consisting of seven prognostic-related genes, including <i>CAV2</i>, <i>EREG</i>, <i>NGFRAP1</i>, <i>WBSCR22</i>, <i>SPINT2</i>, <i>CCDC28A</i>, and <i>BCL10</i>, was constructed and validated. The proficiency and credibility of the signature were verified in both internal and external datasets, and the results showed that the seven-gene signature could effectively predict the prognosis of patients with CC under various clinical conditions. A nomogram was then constructed based on features such as the RiskScore, patients' age, neoplasm stage, and tumor (T), nodes (N), and metastases (M) classification, and the nomogram had good clinical utility. Higher RiskScores were associated with a higher tumour mutational burden, which was confirmed to be a prognostic risk factor. Gene set enrichment analysis showed that high-score groups were enriched in ‘cytoplasmic DNA sensing’, ‘Extracellular matrix receptor interactions’, and ‘focal adhesion’, and low-score groups were enriched in ‘natural killer cell-mediated cytotoxicity’, and ‘T-cell receptor signalling pathways’, among other pathways. A robust seven-gene marker for CC was identified based on scRNA-seq data and was validated in multiple independent cohort studies. These findings provide a new potential marker to predict the prognosis of patients with CC.</p>\",\"PeriodicalId\":50379,\"journal\":{\"name\":\"IET Systems Biology\",\"volume\":\"16 2\",\"pages\":\"72-83\"},\"PeriodicalIF\":1.9000,\"publicationDate\":\"2022-03-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ietresearch.onlinelibrary.wiley.com/doi/epdf/10.1049/syb2.12041\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"IET Systems Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1049/syb2.12041\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"IET Systems Biology","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1049/syb2.12041","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 3
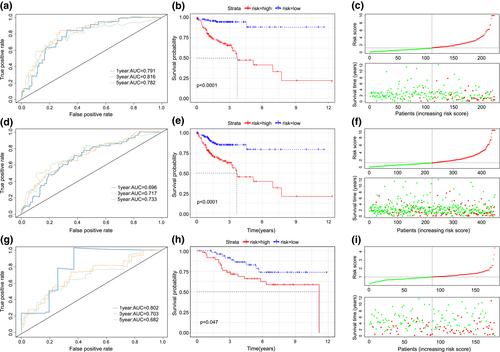
摘要
结肠癌(CC)是世界上最常见的肿瘤之一。单细胞RNA测序(scRNA-seq)能够准确反映肿瘤细胞内及细胞间的异质性,识别与肿瘤发生生长相关的重要基因。在本研究中,scRNA-seq用于鉴定可靠的CC预后生物标志物,首先从基因表达Omnibus数据库下载5-氟尿嘧啶治疗前后CC的scRNA-seq数据。对数据进行预处理,利用主成分分析和t分布随机邻居嵌入算法进行降维。此外,从the Cancer Genome Atlas数据库中获得了CC患者的转录组数据、体细胞变异数据和临床报告。使用Cox回归分析和最小绝对收缩和选择算子方法确定了七个关键基因,以建立与CC预后相关的特征。鉴定的特征在独立的数据集上得到验证,并进一步探索体细胞突变和潜在的致癌途径。基于这些特征、基因特征和其他临床变量,构建了一个更有效的CC患者预测模型nomogram,并进行决策曲线分析来评估nomogram的效用。构建并验证了由CAV2、EREG、NGFRAP1、WBSCR22、SPINT2、CCDC28A和BCL10等7个预后相关基因组成的预后特征。在内部和外部数据集中验证了签名的熟练度和可信度,结果表明,七基因签名可以有效预测CC患者在各种临床条件下的预后。然后根据RiskScore、患者年龄、肿瘤分期、肿瘤(T)、淋巴结(N)和转移(M)分类等特征构建nomogram, nomogram具有良好的临床应用价值。较高的风险评分与较高的肿瘤突变负担相关,这被证实是一个预后风险因素。基因集富集分析显示,高分组富集于“细胞质DNA传感”、“细胞外基质受体相互作用”和“局灶黏附”,而低分组富集于“自然杀伤细胞介导的细胞毒性”和“t细胞受体信号通路”等途径。基于scRNA-seq数据确定了一个强大的七基因CC标记,并在多个独立队列研究中得到验证。这些发现为预测CC患者预后提供了一个新的潜在指标。
Identification and validation of a seven-gene prognostic marker in colon cancer based on single-cell transcriptome analysis
Colon cancer (CC) is one of the most commonly diagnosed tumours worldwide. Single-cell RNA sequencing (scRNA-seq) can accurately reflect the heterogeneity within and between tumour cells and identify important genes associated with cancer development and growth. In this study, scRNA-seq was used to identify reliable prognostic biomarkers in CC. ScRNA-seq data of CC before and after 5-fluorouracil treatment were first downloaded from the Gene Expression Omnibus database. The data were pre-processed, and dimensionality reduction was performed using principal component analysis and t-distributed stochastic neighbour embedding algorithms. Additionally, the transcriptome data, somatic variant data, and clinical reports of patients with CC were obtained from The Cancer Genome Atlas database. Seven key genes were identified using Cox regression analysis and the least absolute shrinkage and selection operator method to establish signatures associated with CC prognoses. The identified signatures were validated on independent datasets, and somatic mutations and potential oncogenic pathways were further explored. Based on these features, gene signatures, and other clinical variables, a more effective predictive model nomogram for patients with CC was constructed, and a decision curve analysis was performed to assess the utility of the nomogram. A prognostic signature consisting of seven prognostic-related genes, including CAV2, EREG, NGFRAP1, WBSCR22, SPINT2, CCDC28A, and BCL10, was constructed and validated. The proficiency and credibility of the signature were verified in both internal and external datasets, and the results showed that the seven-gene signature could effectively predict the prognosis of patients with CC under various clinical conditions. A nomogram was then constructed based on features such as the RiskScore, patients' age, neoplasm stage, and tumor (T), nodes (N), and metastases (M) classification, and the nomogram had good clinical utility. Higher RiskScores were associated with a higher tumour mutational burden, which was confirmed to be a prognostic risk factor. Gene set enrichment analysis showed that high-score groups were enriched in ‘cytoplasmic DNA sensing’, ‘Extracellular matrix receptor interactions’, and ‘focal adhesion’, and low-score groups were enriched in ‘natural killer cell-mediated cytotoxicity’, and ‘T-cell receptor signalling pathways’, among other pathways. A robust seven-gene marker for CC was identified based on scRNA-seq data and was validated in multiple independent cohort studies. These findings provide a new potential marker to predict the prognosis of patients with CC.
期刊介绍:
IET Systems Biology covers intra- and inter-cellular dynamics, using systems- and signal-oriented approaches. Papers that analyse genomic data in order to identify variables and basic relationships between them are considered if the results provide a basis for mathematical modelling and simulation of cellular dynamics. Manuscripts on molecular and cell biological studies are encouraged if the aim is a systems approach to dynamic interactions within and between cells.
The scope includes the following topics:
Genomics, transcriptomics, proteomics, metabolomics, cells, tissue and the physiome; molecular and cellular interaction, gene, cell and protein function; networks and pathways; metabolism and cell signalling; dynamics, regulation and control; systems, signals, and information; experimental data analysis; mathematical modelling, simulation and theoretical analysis; biological modelling, simulation, prediction and control; methodologies, databases, tools and algorithms for modelling and simulation; modelling, analysis and control of biological networks; synthetic biology and bioengineering based on systems biology.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
