Philip J Freda, Attri Ghosh, Elizabeth Zhang, Tianhao Luo, Apurva S Chitre, Oksana Polesskaya, Celine L St Pierre, Jianjun Gao, Connor D Martin, Hao Chen, Angel G Garcia-Martinez, Tengfei Wang, Wenyan Han, Keita Ishiwari, Paul Meyer, Alexander Lamparelli, Christopher P King, Abraham A Palmer, Ruowang Li, Jason H Moore
{"title":"自动数量性状位点分析(AutoQTL)。","authors":"Philip J Freda, Attri Ghosh, Elizabeth Zhang, Tianhao Luo, Apurva S Chitre, Oksana Polesskaya, Celine L St Pierre, Jianjun Gao, Connor D Martin, Hao Chen, Angel G Garcia-Martinez, Tengfei Wang, Wenyan Han, Keita Ishiwari, Paul Meyer, Alexander Lamparelli, Christopher P King, Abraham A Palmer, Ruowang Li, Jason H Moore","doi":"10.1186/s13040-023-00331-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Quantitative Trait Locus (QTL) analysis and Genome-Wide Association Studies (GWAS) have the power to identify variants that capture significant levels of phenotypic variance in complex traits. However, effort and time are required to select the best methods and optimize parameters and pre-processing steps. Although machine learning approaches have been shown to greatly assist in optimization and data processing, applying them to QTL analysis and GWAS is challenging due to the complexity of large, heterogenous datasets. Here, we describe proof-of-concept for an automated machine learning approach, AutoQTL, with the ability to automate many complicated decisions related to analysis of complex traits and generate solutions to describe relationships that exist in genetic data.</p><p><strong>Results: </strong>Using a publicly available dataset of 18 putative QTL from a large-scale GWAS of body mass index in the laboratory rat, Rattus norvegicus, AutoQTL captures the phenotypic variance explained under a standard additive model. AutoQTL also detects evidence of non-additive effects including deviations from additivity and 2-way epistatic interactions in simulated data via multiple optimal solutions. Additionally, feature importance metrics provide different insights into the inheritance models and predictive power of multiple GWAS-derived putative QTL.</p><p><strong>Conclusions: </strong>This proof-of-concept illustrates that automated machine learning techniques can complement standard approaches and have the potential to detect both additive and non-additive effects via various optimal solutions and feature importance metrics. In the future, we aim to expand AutoQTL to accommodate omics-level datasets with intelligent feature selection and feature engineering strategies.</p>","PeriodicalId":48947,"journal":{"name":"Biodata Mining","volume":"16 1","pages":"14"},"PeriodicalIF":6.1000,"publicationDate":"2023-04-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10088184/pdf/","citationCount":"0","resultStr":"{\"title\":\"Automated quantitative trait locus analysis (AutoQTL).\",\"authors\":\"Philip J Freda, Attri Ghosh, Elizabeth Zhang, Tianhao Luo, Apurva S Chitre, Oksana Polesskaya, Celine L St Pierre, Jianjun Gao, Connor D Martin, Hao Chen, Angel G Garcia-Martinez, Tengfei Wang, Wenyan Han, Keita Ishiwari, Paul Meyer, Alexander Lamparelli, Christopher P King, Abraham A Palmer, Ruowang Li, Jason H Moore\",\"doi\":\"10.1186/s13040-023-00331-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Quantitative Trait Locus (QTL) analysis and Genome-Wide Association Studies (GWAS) have the power to identify variants that capture significant levels of phenotypic variance in complex traits. However, effort and time are required to select the best methods and optimize parameters and pre-processing steps. Although machine learning approaches have been shown to greatly assist in optimization and data processing, applying them to QTL analysis and GWAS is challenging due to the complexity of large, heterogenous datasets. Here, we describe proof-of-concept for an automated machine learning approach, AutoQTL, with the ability to automate many complicated decisions related to analysis of complex traits and generate solutions to describe relationships that exist in genetic data.</p><p><strong>Results: </strong>Using a publicly available dataset of 18 putative QTL from a large-scale GWAS of body mass index in the laboratory rat, Rattus norvegicus, AutoQTL captures the phenotypic variance explained under a standard additive model. AutoQTL also detects evidence of non-additive effects including deviations from additivity and 2-way epistatic interactions in simulated data via multiple optimal solutions. Additionally, feature importance metrics provide different insights into the inheritance models and predictive power of multiple GWAS-derived putative QTL.</p><p><strong>Conclusions: </strong>This proof-of-concept illustrates that automated machine learning techniques can complement standard approaches and have the potential to detect both additive and non-additive effects via various optimal solutions and feature importance metrics. In the future, we aim to expand AutoQTL to accommodate omics-level datasets with intelligent feature selection and feature engineering strategies.</p>\",\"PeriodicalId\":48947,\"journal\":{\"name\":\"Biodata Mining\",\"volume\":\"16 1\",\"pages\":\"14\"},\"PeriodicalIF\":6.1000,\"publicationDate\":\"2023-04-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10088184/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biodata Mining\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13040-023-00331-3\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biodata Mining","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13040-023-00331-3","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Automated quantitative trait locus analysis (AutoQTL).
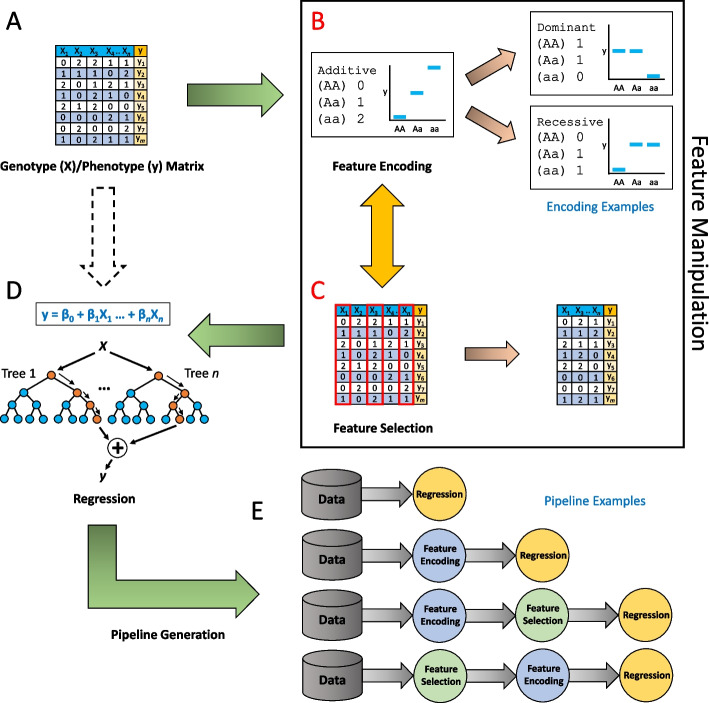
Background: Quantitative Trait Locus (QTL) analysis and Genome-Wide Association Studies (GWAS) have the power to identify variants that capture significant levels of phenotypic variance in complex traits. However, effort and time are required to select the best methods and optimize parameters and pre-processing steps. Although machine learning approaches have been shown to greatly assist in optimization and data processing, applying them to QTL analysis and GWAS is challenging due to the complexity of large, heterogenous datasets. Here, we describe proof-of-concept for an automated machine learning approach, AutoQTL, with the ability to automate many complicated decisions related to analysis of complex traits and generate solutions to describe relationships that exist in genetic data.
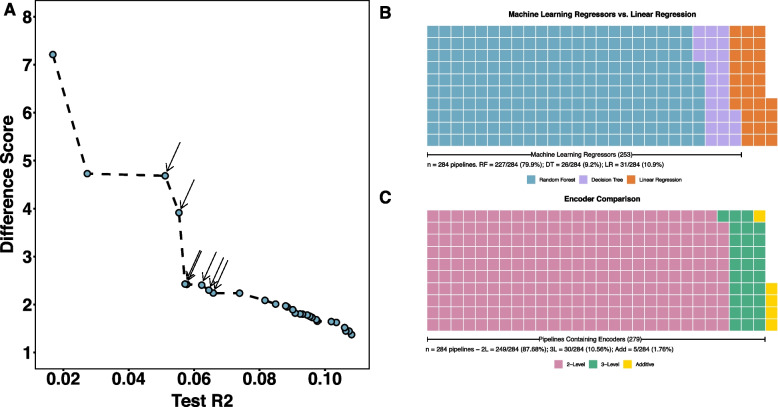
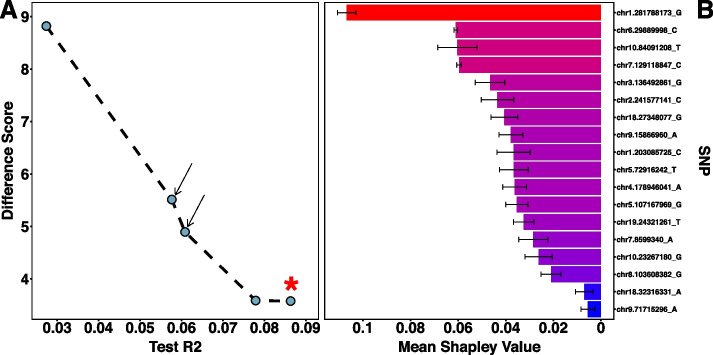
Results: Using a publicly available dataset of 18 putative QTL from a large-scale GWAS of body mass index in the laboratory rat, Rattus norvegicus, AutoQTL captures the phenotypic variance explained under a standard additive model. AutoQTL also detects evidence of non-additive effects including deviations from additivity and 2-way epistatic interactions in simulated data via multiple optimal solutions. Additionally, feature importance metrics provide different insights into the inheritance models and predictive power of multiple GWAS-derived putative QTL.
Conclusions: This proof-of-concept illustrates that automated machine learning techniques can complement standard approaches and have the potential to detect both additive and non-additive effects via various optimal solutions and feature importance metrics. In the future, we aim to expand AutoQTL to accommodate omics-level datasets with intelligent feature selection and feature engineering strategies.
期刊介绍:
BioData Mining is an open access, open peer-reviewed journal encompassing research on all aspects of data mining applied to high-dimensional biological and biomedical data, focusing on computational aspects of knowledge discovery from large-scale genetic, transcriptomic, genomic, proteomic, and metabolomic data.
Topical areas include, but are not limited to:
-Development, evaluation, and application of novel data mining and machine learning algorithms.
-Adaptation, evaluation, and application of traditional data mining and machine learning algorithms.
-Open-source software for the application of data mining and machine learning algorithms.
-Design, development and integration of databases, software and web services for the storage, management, retrieval, and analysis of data from large scale studies.
-Pre-processing, post-processing, modeling, and interpretation of data mining and machine learning results for biological interpretation and knowledge discovery.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
