{"title":"UBR7 E3 Ligase Suppresses Interferon-β Mediated Immune Signaling by Targeting Sp110 in Hepatitis B Virus-Induced Hepatocellular Carcinoma.","authors":"Vipin Singh, Atanu Mondal, Santanu Adhikary, Payel Mondal, Niranjan Shirgaonkar, Ramanuj DasGupta, Siddhartha Roy, Chandrima Das","doi":"10.1021/acsinfecdis.4c00213","DOIUrl":null,"url":null,"abstract":"<p><p>A newly discovered E3 ubiquitin ligase, UBR7, plays a crucial role in histone H2BK120 monoubiquitination. Here, we report a novel function of UBR7 in promoting hepatitis B virus (HBV) pathogenesis, which further leads to HBV-induced hepatocellular carcinoma (HCC). Transcriptomics analysis from HCC patients revealed the deregulation of UBR7 in cancer. Remarkably, targeting UBR7, particularly its catalytic function, led to a significant decrease in viral copy numbers. We also identified the speckled family protein Sp110 as an important substrate of UBR7. Notably, Sp110 has been previously shown to be a resident of promyelocytic leukemia nuclear bodies (PML-NBs), where it remains SUMOylated, and during HBV infection, it undergoes deSUMOylation and exits the PML body. We observed that UBR7 ubiquitinates Sp110 at critical residues within its SAND domain. Sp110 ubiquitination downregulates genes in the type I interferon response pathway. Comparative analysis of RNA-Seq from the UBR7/Sp110 knockdown data set confirmed that the IFN-β signaling pathway gets deregulated in HCC cells in the presence of HBV. Single-cell RNA-Seq analysis of patient samples further confirmed the inverse correlation between the expression of Sp110/UBR7 and the inflammation score. Notably, silencing of UBR7 induces IRF7 phosphorylation, thereby augmenting interferon (IFN)-β and the downstream interferon-stimulated genes (ISGs). Further, wild-type but not the ubiquitination-defective mutant of Sp110 could be recruited to the type I interferon response pathway genes. Our study establishes a new function of UBR7 in non-histone protein ubiquitination, promoting viral persistence, and has important implications for the development of therapeutic strategies targeting HBV-induced HCC.</p>","PeriodicalId":17,"journal":{"name":"ACS Infectious Diseases","volume":" ","pages":"3775-3796"},"PeriodicalIF":3.8000,"publicationDate":"2024-11-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Infectious Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1021/acsinfecdis.4c00213","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/6/28 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
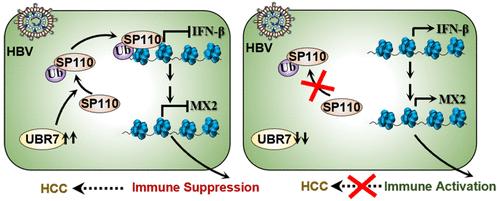
Abstract
A newly discovered E3 ubiquitin ligase, UBR7, plays a crucial role in histone H2BK120 monoubiquitination. Here, we report a novel function of UBR7 in promoting hepatitis B virus (HBV) pathogenesis, which further leads to HBV-induced hepatocellular carcinoma (HCC). Transcriptomics analysis from HCC patients revealed the deregulation of UBR7 in cancer. Remarkably, targeting UBR7, particularly its catalytic function, led to a significant decrease in viral copy numbers. We also identified the speckled family protein Sp110 as an important substrate of UBR7. Notably, Sp110 has been previously shown to be a resident of promyelocytic leukemia nuclear bodies (PML-NBs), where it remains SUMOylated, and during HBV infection, it undergoes deSUMOylation and exits the PML body. We observed that UBR7 ubiquitinates Sp110 at critical residues within its SAND domain. Sp110 ubiquitination downregulates genes in the type I interferon response pathway. Comparative analysis of RNA-Seq from the UBR7/Sp110 knockdown data set confirmed that the IFN-β signaling pathway gets deregulated in HCC cells in the presence of HBV. Single-cell RNA-Seq analysis of patient samples further confirmed the inverse correlation between the expression of Sp110/UBR7 and the inflammation score. Notably, silencing of UBR7 induces IRF7 phosphorylation, thereby augmenting interferon (IFN)-β and the downstream interferon-stimulated genes (ISGs). Further, wild-type but not the ubiquitination-defective mutant of Sp110 could be recruited to the type I interferon response pathway genes. Our study establishes a new function of UBR7 in non-histone protein ubiquitination, promoting viral persistence, and has important implications for the development of therapeutic strategies targeting HBV-induced HCC.
期刊介绍:
ACS Infectious Diseases will be the first journal to highlight chemistry and its role in this multidisciplinary and collaborative research area. The journal will cover a diverse array of topics including, but not limited to:
* Discovery and development of new antimicrobial agents — identified through target- or phenotypic-based approaches as well as compounds that induce synergy with antimicrobials.
* Characterization and validation of drug target or pathways — use of single target and genome-wide knockdown and knockouts, biochemical studies, structural biology, new technologies to facilitate characterization and prioritization of potential drug targets.
* Mechanism of drug resistance — fundamental research that advances our understanding of resistance; strategies to prevent resistance.
* Mechanisms of action — use of genetic, metabolomic, and activity- and affinity-based protein profiling to elucidate the mechanism of action of clinical and experimental antimicrobial agents.
* Host-pathogen interactions — tools for studying host-pathogen interactions, cellular biochemistry of hosts and pathogens, and molecular interactions of pathogens with host microbiota.
* Small molecule vaccine adjuvants for infectious disease.
* Viral and bacterial biochemistry and molecular biology.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
