All-body concept and quantified limits of cooperativity and related effects in homodromic cyclic water clusters from a molecular-wide and electron density-based approach
Ignacy Cukrowski, Stéfan Zaaiman, Shahnawaz Hussain, Jurgens H. de Lange
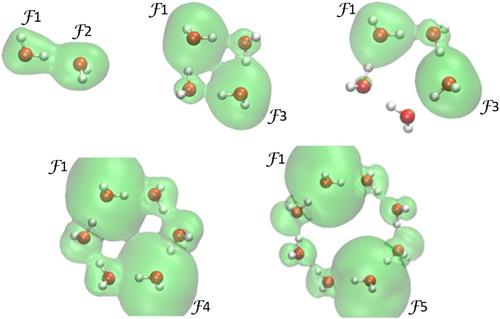
{"title":"All-body concept and quantified limits of cooperativity and related effects in homodromic cyclic water clusters from a molecular-wide and electron density-based approach","authors":"Ignacy Cukrowski, Stéfan Zaaiman, Shahnawaz Hussain, Jurgens H. de Lange","doi":"10.1002/jcc.27489","DOIUrl":null,"url":null,"abstract":"<p>We strongly advocate distinguishing cooperativity from cooperativity-induced effects. From the MOW<i>e</i>D-based approach, the origin of <b>all-body</b> cooperativity is synonymous with physics- and quantum-based processes of electron (<i>e</i>) delocalization throughout water clusters. To this effect, over 10 atom-pairs contribute to the total <i>e</i>-density at a BCP(H,O) between water molecules in a tetramer. Intermolecular all-body <i>e</i>-delocalization, that is, cooperativity, is an energy-minimizing process that fully explains non-additive increase in stability of a water molecule in clusters with an increase in their size. A non-linear change in cooperativity and cooperativity-induced effects, such as (i) structural (e.g., a change in d(O,O)) or topological intra- and intermolecular properties in water clusters (e.g., electron density or potential energy density at bond critical points) is theoretically reproduced by the proposed expression. It predicted the limiting value of delocalized electrons by a H<sub>2</sub>O molecule in homodromic cyclic clusters to be 1.58<i>e</i>. O-atoms provide the vast majority of electrons that “travel throughout a cluster predominantly on a <i>privileged</i> exchange quantum density highway” (⋅⋅⋅O–H⋅⋅⋅O–H⋅⋅⋅O–H⋅⋅⋅) using Bader's classical bond paths as density bridges linking water molecules. There are, however, <i>additional</i> electron exchange channels that are not seen on molecular graphs as bond paths. A 3D visual representation of the “privileged” and “additional” exchange channels as well as detailed intra- and inter-molecular patterns of <i>e</i>-sharing and (de)localizing is presented. The energy stabilizing contribution made by three O-atoms of neighboring water molecules was found to be large (−597 kcal/mol in cyclic hexamer) and 5 times more significant than that of a classical O–H⋅⋅⋅O intermolecular H-bond.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 32","pages":"2812-2824"},"PeriodicalIF":4.8000,"publicationDate":"2024-08-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27489","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27489","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
We strongly advocate distinguishing cooperativity from cooperativity-induced effects. From the MOWeD-based approach, the origin of all-body cooperativity is synonymous with physics- and quantum-based processes of electron (e) delocalization throughout water clusters. To this effect, over 10 atom-pairs contribute to the total e-density at a BCP(H,O) between water molecules in a tetramer. Intermolecular all-body e-delocalization, that is, cooperativity, is an energy-minimizing process that fully explains non-additive increase in stability of a water molecule in clusters with an increase in their size. A non-linear change in cooperativity and cooperativity-induced effects, such as (i) structural (e.g., a change in d(O,O)) or topological intra- and intermolecular properties in water clusters (e.g., electron density or potential energy density at bond critical points) is theoretically reproduced by the proposed expression. It predicted the limiting value of delocalized electrons by a H2O molecule in homodromic cyclic clusters to be 1.58e. O-atoms provide the vast majority of electrons that “travel throughout a cluster predominantly on a privileged exchange quantum density highway” (⋅⋅⋅O–H⋅⋅⋅O–H⋅⋅⋅O–H⋅⋅⋅) using Bader's classical bond paths as density bridges linking water molecules. There are, however, additional electron exchange channels that are not seen on molecular graphs as bond paths. A 3D visual representation of the “privileged” and “additional” exchange channels as well as detailed intra- and inter-molecular patterns of e-sharing and (de)localizing is presented. The energy stabilizing contribution made by three O-atoms of neighboring water molecules was found to be large (−597 kcal/mol in cyclic hexamer) and 5 times more significant than that of a classical O–H⋅⋅⋅O intermolecular H-bond.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
