Jordan Howe, Salsabil Abou-Hatab, Spiridoula Matsika
{"title":"Modeling the effect of substituents on the electronically excited states of indole derivatives","authors":"Jordan Howe, Salsabil Abou-Hatab, Spiridoula Matsika","doi":"10.1002/jcc.27502","DOIUrl":null,"url":null,"abstract":"<p>A proper understanding of excited state properties of indole derivatives can lead to rational design of efficient fluorescent probes. The optically active <span></span><math>\n <mrow>\n <msub>\n <mrow>\n <mi>L</mi>\n </mrow>\n <mrow>\n <mi>a</mi>\n </mrow>\n </msub>\n </mrow></math> and <span></span><math>\n <mrow>\n <msub>\n <mrow>\n <mi>L</mi>\n </mrow>\n <mrow>\n <mi>b</mi>\n </mrow>\n </msub>\n </mrow></math> excited states of a series of substituted indoles, where a substituent was placed on position four, were calculated using equation of motion coupled cluster and time dependent density functional theory. The results indicate that most substituted indoles have a brighter second excited state corresponding to experimental absorption maxima, but a few with electron withdrawing substituents absorb more on the first excited state. Absorption on the first excited state may increase their fluorescence quantum yield, making them better probes. Electronic structure methods were found to predict the energies of the systems with electron withdrawing substituents more accurately than those with electron donating substituents. The excited states of both states correlated well with electrophilicity, similar to the experimental trends for the absorption maxima. Overall, these computational studies indicate that theory can be used to predict excited state properties of substituted indoles, when the substituent is an electron withdrawing group.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2024-09-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27502","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
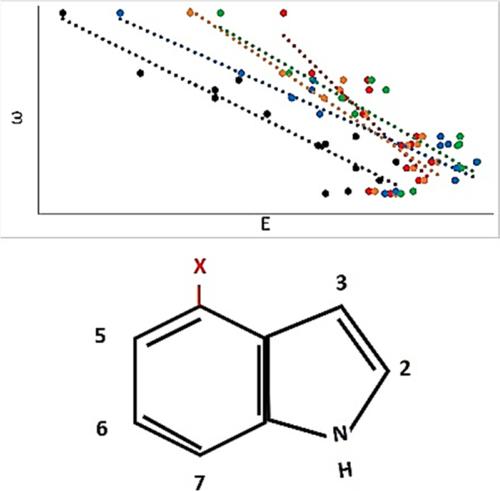
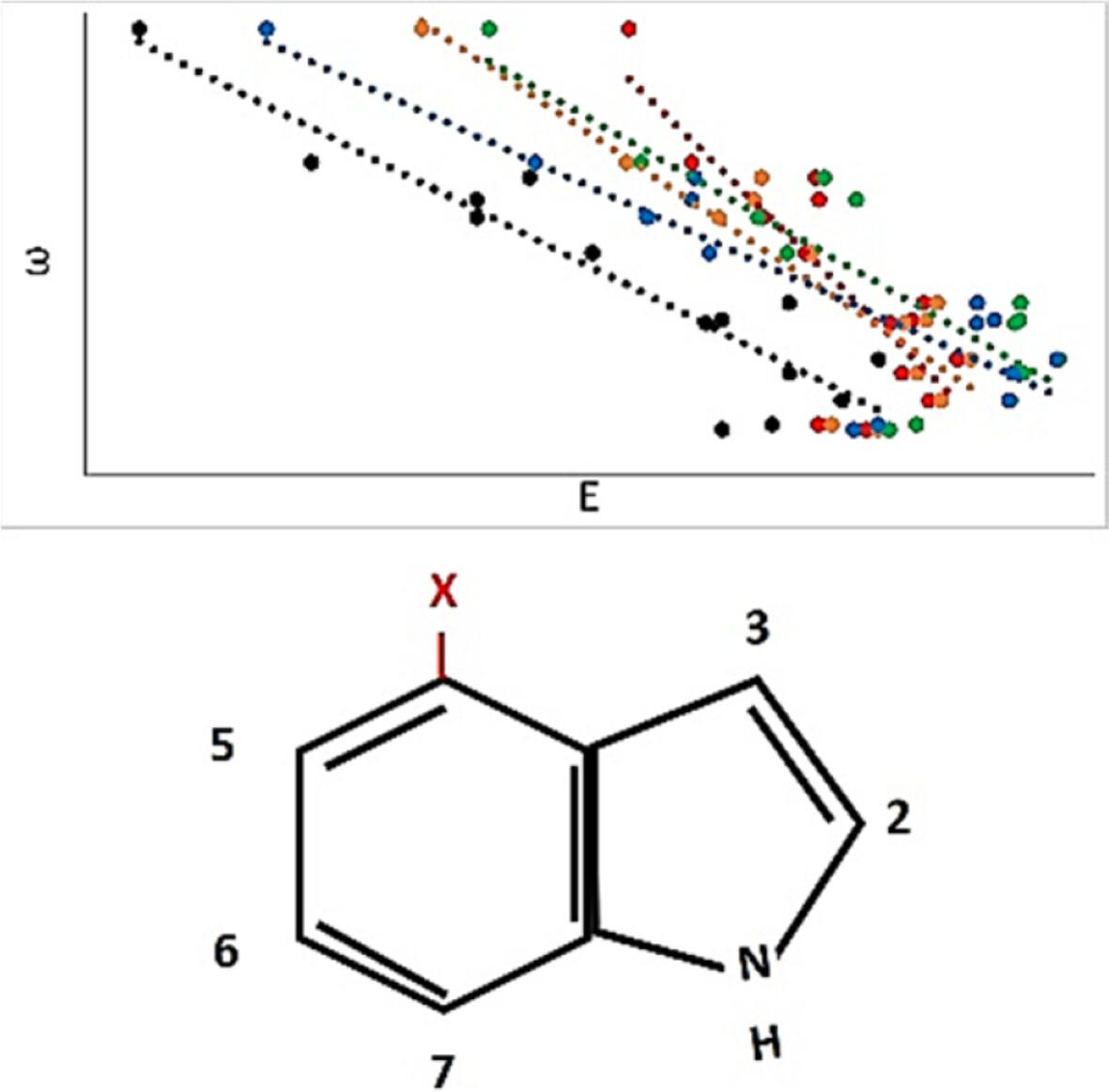
A proper understanding of excited state properties of indole derivatives can lead to rational design of efficient fluorescent probes. The optically active and excited states of a series of substituted indoles, where a substituent was placed on position four, were calculated using equation of motion coupled cluster and time dependent density functional theory. The results indicate that most substituted indoles have a brighter second excited state corresponding to experimental absorption maxima, but a few with electron withdrawing substituents absorb more on the first excited state. Absorption on the first excited state may increase their fluorescence quantum yield, making them better probes. Electronic structure methods were found to predict the energies of the systems with electron withdrawing substituents more accurately than those with electron donating substituents. The excited states of both states correlated well with electrophilicity, similar to the experimental trends for the absorption maxima. Overall, these computational studies indicate that theory can be used to predict excited state properties of substituted indoles, when the substituent is an electron withdrawing group.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
