{"title":"Fluxional halogen bonds in linear complexes of tetrafluorodiiodobenzene with dinitrobenzene","authors":"Cai-Yue Gao, Bin-Bin Pei, Si-Dian Li","doi":"10.1002/jcc.27483","DOIUrl":null,"url":null,"abstract":"<p>The fluxional nature of halogen bonds (XBs) in small molecular clusters, supramolecules, and molecular crystals has received considerable attention in recent years. In this work, based on extensive density-functional theory calculations and detailed electrostatic potential (ESP), natural bonding orbital (NBO), non-covalent interactions-reduced density gradient (NCI-RDG), and quantum theory of atoms in molecules (QTAIM) analyses, we unveil the existence of fluxional halogen bonds (FXBs) in a series of linear (IC<sub>6</sub>F<sub>4</sub>I)<sub><i>m</i></sub>(OONC<sub>6</sub>H<sub>4</sub>NOO)<sub><i>n</i></sub> (<i>m</i> + <i>n</i> = 2–5) complexes of tetrafluorodiiodobenzene with dinitrobenzene which appear to be similar to the previously reported fluxional hydrogen bonds (FHBs) in small water clusters (H<sub>2</sub>O)<sub><i>n</i></sub> (<i>n</i> = 2–6). The obtained <span></span><math>\n <mrow>\n <mi>GS</mi>\n <mo>⇌</mo>\n <mi>TS</mi>\n <mo>⇌</mo>\n <msup>\n <mi>GS</mi>\n <mo>'</mo>\n </msup>\n </mrow></math> fluxional mechanisms involve one FXB in the systems which fluctuates reversibly between two linear C<span></span>I···O XBs in the ground states (GS and GS') via a bifurcated C<span></span>I O<sub>2</sub>N van der Waals interaction in the transition state (TS). The cohesive energies (<i>E</i><sub>coh</sub>) of these complexes with up to four XBs exhibit an almost perfect linear relationship with the numbers of XBs in the systems, with the average calculated halogen bond energy of <i>E</i><sub>coh/XB</sub> = 3.48 kcal·mol<sup>−1</sup> in the ground states which appears to be about 55% of the average calculated hydrogen bond energy (<i>E</i><sub>coh/HB</sub> = 6.28 kcal·mol<sup>−1</sup>) in small water clusters.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27483","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
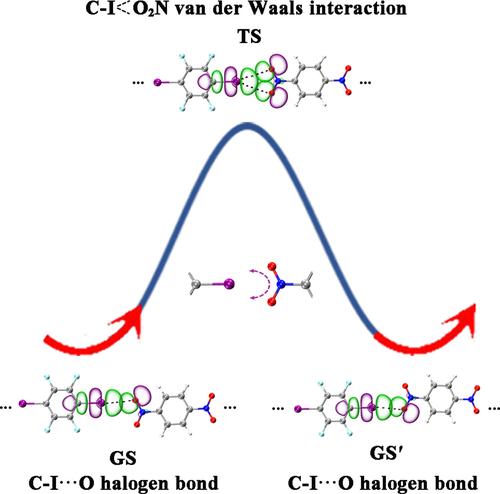
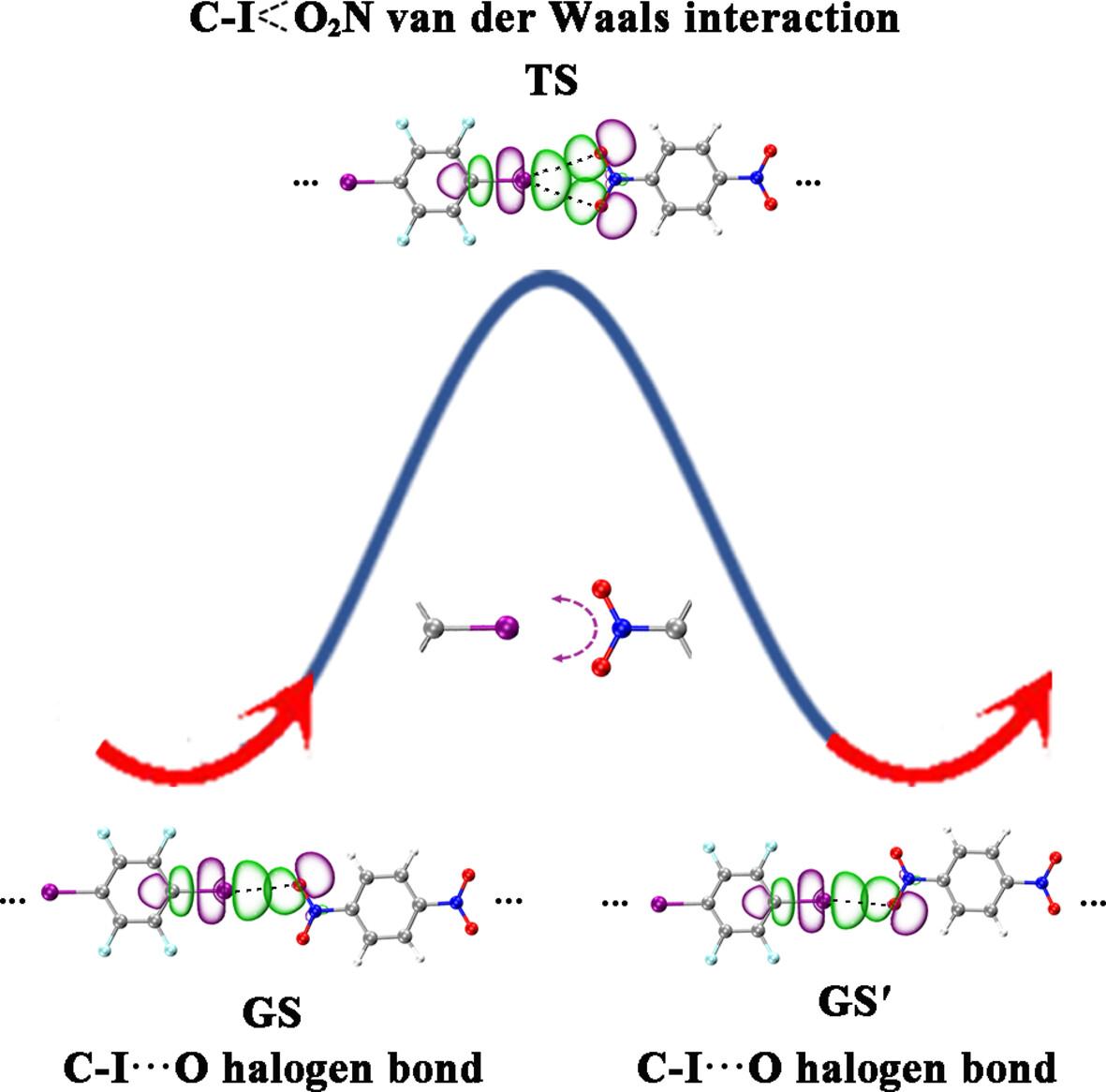
The fluxional nature of halogen bonds (XBs) in small molecular clusters, supramolecules, and molecular crystals has received considerable attention in recent years. In this work, based on extensive density-functional theory calculations and detailed electrostatic potential (ESP), natural bonding orbital (NBO), non-covalent interactions-reduced density gradient (NCI-RDG), and quantum theory of atoms in molecules (QTAIM) analyses, we unveil the existence of fluxional halogen bonds (FXBs) in a series of linear (IC6F4I)m(OONC6H4NOO)n (m + n = 2–5) complexes of tetrafluorodiiodobenzene with dinitrobenzene which appear to be similar to the previously reported fluxional hydrogen bonds (FHBs) in small water clusters (H2O)n (n = 2–6). The obtained fluxional mechanisms involve one FXB in the systems which fluctuates reversibly between two linear CI···O XBs in the ground states (GS and GS') via a bifurcated CI O2N van der Waals interaction in the transition state (TS). The cohesive energies (Ecoh) of these complexes with up to four XBs exhibit an almost perfect linear relationship with the numbers of XBs in the systems, with the average calculated halogen bond energy of Ecoh/XB = 3.48 kcal·mol−1 in the ground states which appears to be about 55% of the average calculated hydrogen bond energy (Ecoh/HB = 6.28 kcal·mol−1) in small water clusters.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
