Rodrigo Arcoverde Cerveira , Klara Lenart , Marcel Martin , Matthew James Hinchcliff , Fredrika Hellgren , Kewei Ye , Juliana Assis Geraldo , Taras Kreslavsky , Sebastian Ols , Karin Loré
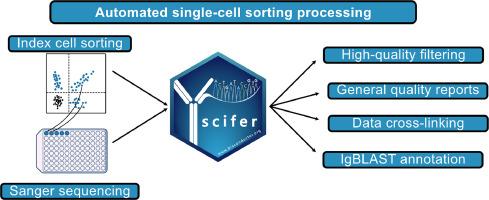
{"title":"Scifer: An R/Bioconductor package for large-scale integration of Sanger sequencing and flow cytometry data of index-sorted single cells","authors":"Rodrigo Arcoverde Cerveira , Klara Lenart , Marcel Martin , Matthew James Hinchcliff , Fredrika Hellgren , Kewei Ye , Juliana Assis Geraldo , Taras Kreslavsky , Sebastian Ols , Karin Loré","doi":"10.1016/j.immuno.2024.100046","DOIUrl":null,"url":null,"abstract":"<div><div>Sanger sequencing remains widely used in various experimental contexts, often in combination with flow cytometry for indexing specific cell populations. However, existing software lacks the capability to automate quality control (QC) of raw Sanger sequencing data and integrate it with flow cytometry information on a large scale. Here, we introduce scifer, an R package now available in the latest release of Bioconductor (3.20) showcasing its effectiveness in seamlessly integrating these types of data as demonstrated by analyses of B cell and T cell receptor sequences. Scifer preprocesses raw data from index sorts and immune receptor Sanger sequencing. It identifies high-quality sequences based on selected parameters, such as length, Phred scores, and heavy-chain complementarity-determining region 3 (HCDR3) quality. As a result, the quality of germline assignments is significantly increased and spurious variable gene mutations are reduced. Scifer is automated and can process thousands of sequences in less than an hour. Its output provides quality control reports, FASTA files, summarized tables, and electropherograms for manual inspection. In summary, scifer is a user-friendly software that speeds up the analysis of immune receptor repertoire sequences, offering wide applicability.</div></div>","PeriodicalId":73343,"journal":{"name":"Immunoinformatics (Amsterdam, Netherlands)","volume":"16 ","pages":"Article 100046"},"PeriodicalIF":0.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Immunoinformatics (Amsterdam, Netherlands)","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2667119024000168","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/29 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Sanger sequencing remains widely used in various experimental contexts, often in combination with flow cytometry for indexing specific cell populations. However, existing software lacks the capability to automate quality control (QC) of raw Sanger sequencing data and integrate it with flow cytometry information on a large scale. Here, we introduce scifer, an R package now available in the latest release of Bioconductor (3.20) showcasing its effectiveness in seamlessly integrating these types of data as demonstrated by analyses of B cell and T cell receptor sequences. Scifer preprocesses raw data from index sorts and immune receptor Sanger sequencing. It identifies high-quality sequences based on selected parameters, such as length, Phred scores, and heavy-chain complementarity-determining region 3 (HCDR3) quality. As a result, the quality of germline assignments is significantly increased and spurious variable gene mutations are reduced. Scifer is automated and can process thousands of sequences in less than an hour. Its output provides quality control reports, FASTA files, summarized tables, and electropherograms for manual inspection. In summary, scifer is a user-friendly software that speeds up the analysis of immune receptor repertoire sequences, offering wide applicability.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
