Christopher D. Woodgate, George A. Marchant, Livia B. Pártay, Julie B. Staunton
{"title":"Structure, short-range order, and phase stability of the AlxCrFeCoNi high-entropy alloy: insights from a perturbative, DFT-based analysis","authors":"Christopher D. Woodgate, George A. Marchant, Livia B. Pártay, Julie B. Staunton","doi":"10.1038/s41524-024-01445-w","DOIUrl":null,"url":null,"abstract":"<p>We study the phase behaviour of the Al<sub><i>x</i></sub>CrFeCoNi high-entropy alloy. Our approach is based on a perturbative analysis of the internal energy of the paramagnetic solid solution as evaluated within the Korringa-Kohn-Rostoker formulation of density functional theory, using the coherent potential approximation to average over disorder. Via application of a Landau-type linear response theory, we infer preferential chemical orderings directly. In addition, we recover a pairwise form of the alloy internal energy suitable for study via atomistic simulations, which in this work are performed using the nested sampling algorithm, which is well-suited for studying complex potential energy surfaces. When the underlying lattice is fcc, at low concentrations of Al, depending on the value of <i>x</i>, we predict either an L1<sub>2</sub> or D0<sub>22</sub> ordering emerging below approximately 1000 K. On the other hand, when the underlying lattice is bcc, consistent with experimental observations, we predict B2 ordering temperatures higher than the melting temperature of the alloy, confirming that this ordered phase forms directly from the melt. For both fcc and bcc systems, chemical orderings are dominated by Al moving to one sublattice, Ni and Co the other, while Cr and Fe remain comparatively disordered. On the bcc lattice, our atomistic modelling suggests eventual decomposition into B2 NiAl and Cr-rich phases. These results shed light on the fundamental physical origins of atomic ordering tendencies in these intriguing materials.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"260 1","pages":""},"PeriodicalIF":13.1000,"publicationDate":"2024-11-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01445-w","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
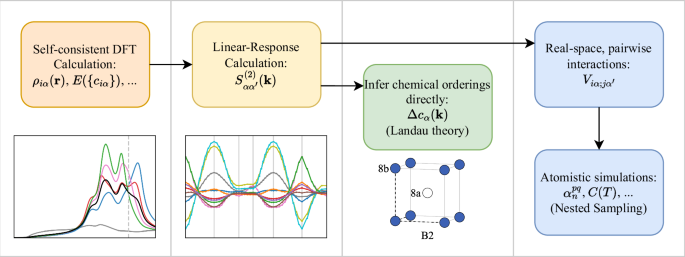
We study the phase behaviour of the AlxCrFeCoNi high-entropy alloy. Our approach is based on a perturbative analysis of the internal energy of the paramagnetic solid solution as evaluated within the Korringa-Kohn-Rostoker formulation of density functional theory, using the coherent potential approximation to average over disorder. Via application of a Landau-type linear response theory, we infer preferential chemical orderings directly. In addition, we recover a pairwise form of the alloy internal energy suitable for study via atomistic simulations, which in this work are performed using the nested sampling algorithm, which is well-suited for studying complex potential energy surfaces. When the underlying lattice is fcc, at low concentrations of Al, depending on the value of x, we predict either an L12 or D022 ordering emerging below approximately 1000 K. On the other hand, when the underlying lattice is bcc, consistent with experimental observations, we predict B2 ordering temperatures higher than the melting temperature of the alloy, confirming that this ordered phase forms directly from the melt. For both fcc and bcc systems, chemical orderings are dominated by Al moving to one sublattice, Ni and Co the other, while Cr and Fe remain comparatively disordered. On the bcc lattice, our atomistic modelling suggests eventual decomposition into B2 NiAl and Cr-rich phases. These results shed light on the fundamental physical origins of atomic ordering tendencies in these intriguing materials.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
