{"title":"Prediction of miRNA-disease associations based on PCA and cascade forest.","authors":"Chuanlei Zhang, Yubo Li, Yinglun Dong, Wei Chen, Changqing Yu","doi":"10.1186/s12859-024-05999-w","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>As a key non-coding RNA molecule, miRNA profoundly affects gene expression regulation and connects to the pathological processes of several kinds of human diseases. However, conventional experimental methods for validating miRNA-disease associations are laborious. Consequently, the development of efficient and reliable computational prediction models is crucial for the identification and validation of these associations.</p><p><strong>Results: </strong>In this research, we developed the PCACFMDA method to predict the potential associations between miRNAs and diseases. To construct a multidimensional feature matrix, we consider the fusion similarities of miRNA and disease and miRNA-disease pairs. We then use principal component analysis(PCA) to reduce data complexity and extract low-dimensional features. Subsequently, a tuned cascade forest is used to mine the features and output prediction scores deeply. The results of the 5-fold cross-validation using the HMDD v2.0 database indicate that the PCACFMDA algorithm achieved an AUC of 98.56%. Additionally, we perform case studies on breast, esophageal and lung neoplasms. The findings revealed that the top 50 miRNAs most strongly linked to each disease have been validated.</p><p><strong>Conclusions: </strong>Based on PCA and optimized cascade forests, we propose the PCACFMDA model for predicting undiscovered miRNA-disease associations. The experimental results demonstrate superior prediction performance and commendable stability. Consequently, the PCACFMDA is a potent instrument for in-depth exploration of miRNA-disease associations.</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"25 1","pages":"386"},"PeriodicalIF":3.3000,"publicationDate":"2024-12-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11660965/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-024-05999-w","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: As a key non-coding RNA molecule, miRNA profoundly affects gene expression regulation and connects to the pathological processes of several kinds of human diseases. However, conventional experimental methods for validating miRNA-disease associations are laborious. Consequently, the development of efficient and reliable computational prediction models is crucial for the identification and validation of these associations.
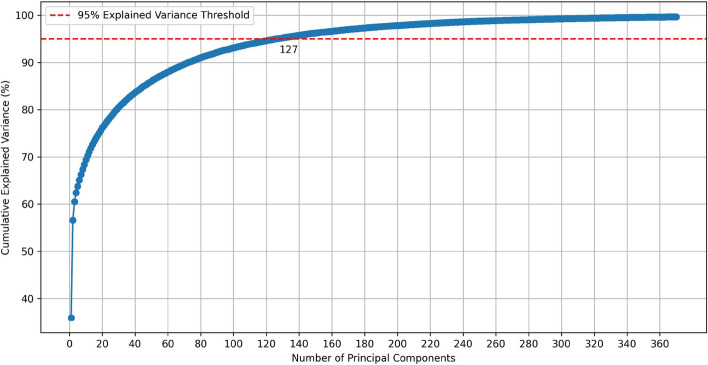
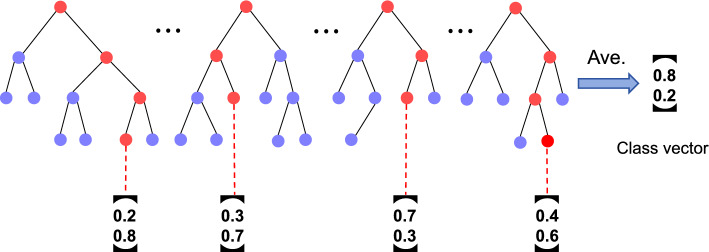
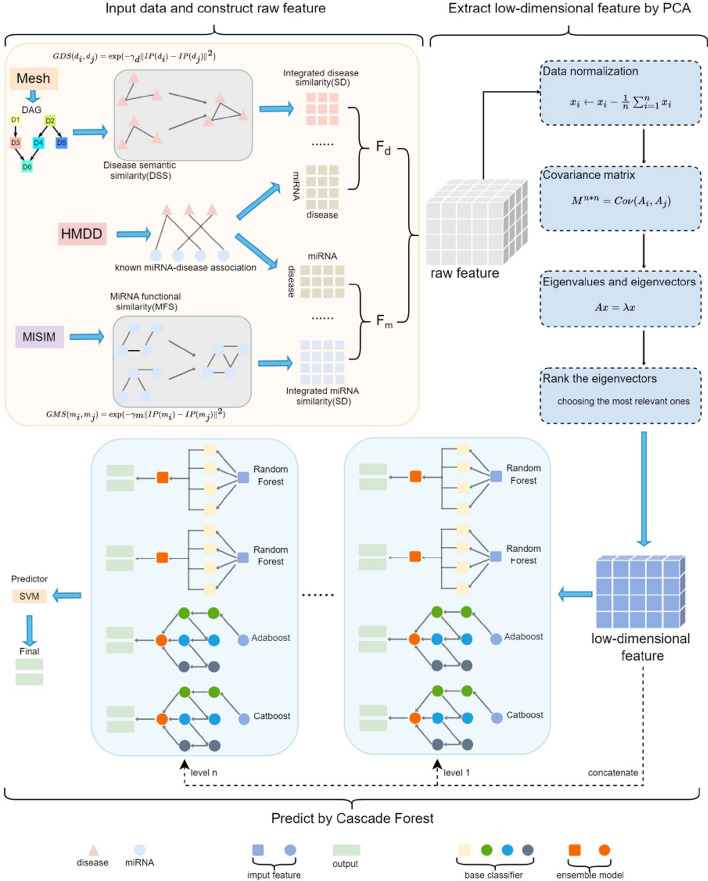
Results: In this research, we developed the PCACFMDA method to predict the potential associations between miRNAs and diseases. To construct a multidimensional feature matrix, we consider the fusion similarities of miRNA and disease and miRNA-disease pairs. We then use principal component analysis(PCA) to reduce data complexity and extract low-dimensional features. Subsequently, a tuned cascade forest is used to mine the features and output prediction scores deeply. The results of the 5-fold cross-validation using the HMDD v2.0 database indicate that the PCACFMDA algorithm achieved an AUC of 98.56%. Additionally, we perform case studies on breast, esophageal and lung neoplasms. The findings revealed that the top 50 miRNAs most strongly linked to each disease have been validated.
Conclusions: Based on PCA and optimized cascade forests, we propose the PCACFMDA model for predicting undiscovered miRNA-disease associations. The experimental results demonstrate superior prediction performance and commendable stability. Consequently, the PCACFMDA is a potent instrument for in-depth exploration of miRNA-disease associations.
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
