Hao Bai, Yanghua He, Yi Ding, Qin Chu, Ling Lian, Eliyahu M Heifetz, Ning Yang, Hans H Cheng, Huanmin Zhang, Jilan Chen, Jiuzhou Song
{"title":"Genome-wide characterization of copy number variations in the host genome in genetic resistance to Marek's disease using next generation sequencing.","authors":"Hao Bai, Yanghua He, Yi Ding, Qin Chu, Ling Lian, Eliyahu M Heifetz, Ning Yang, Hans H Cheng, Huanmin Zhang, Jilan Chen, Jiuzhou Song","doi":"10.1186/s12863-020-00884-w","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Marek's disease (MD) is a highly neoplastic disease primarily affecting chickens, and remains as a chronic infectious disease that threatens the poultry industry. Copy number variation (CNV) has been examined in many species and is recognized as a major source of genetic variation that directly contributes to phenotypic variation such as resistance to infectious diseases. Two highly inbred chicken lines, 6<sub>3</sub> (MD-resistant) and 7<sub>2</sub> (MD-susceptible), as well as their F<sub>1</sub> generation and six recombinant congenic strains (RCSs) with varied susceptibility to MD, are considered as ideal models to identify the complex mechanisms of genetic and molecular resistance to MD.</p><p><strong>Results: </strong>In the present study, to unravel the potential genetic mechanisms underlying resistance to MD, we performed a genome-wide CNV detection using next generation sequencing on the inbred chicken lines with the assistance of CNVnator. As a result, a total of 1649 CNV regions (CNVRs) were successfully identified after merging all the nine datasets, of which 90 CNVRs were overlapped across all the chicken lines. Within these shared regions, 1360 harbored genes were identified. In addition, 55 and 44 CNVRs with 62 and 57 harbored genes were specifically identified in line 6<sub>3</sub> and 7<sub>2</sub>, respectively. Bioinformatics analysis showed that the nearby genes were significantly enriched in 36 GO terms and 6 KEGG pathways including JAK/STAT signaling pathway. Ten CNVRs (nine deletions and one duplication) involved in 10 disease-related genes were selected for validation by using quantitative real-time PCR (qPCR), all of which were successfully confirmed. Finally, qPCR was also used to validate two deletion events in line 7<sub>2</sub> that were definitely normal in line 6<sub>3</sub>. One high-confidence gene, IRF2 was identified as the most promising candidate gene underlying resistance and susceptibility to MD in view of its function and overlaps with data from previous study.</p><p><strong>Conclusions: </strong>Our findings provide valuable insights for understanding the genetic mechanism of resistance to MD and the identified gene and pathway could be considered as the subject of further functional characterization.</p>","PeriodicalId":9197,"journal":{"name":"BMC Genetics","volume":" ","pages":"77"},"PeriodicalIF":2.9000,"publicationDate":"2020-07-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12863-020-00884-w","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s12863-020-00884-w","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 1
Abstract
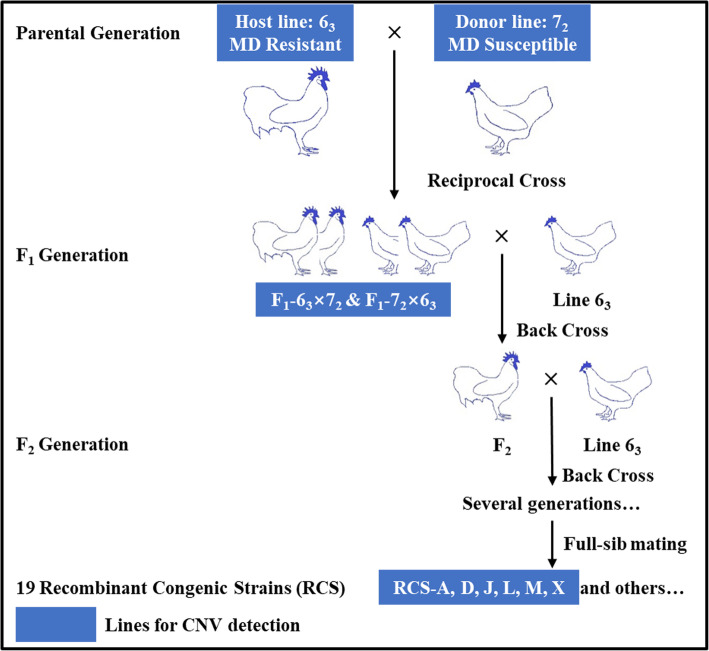
Background: Marek's disease (MD) is a highly neoplastic disease primarily affecting chickens, and remains as a chronic infectious disease that threatens the poultry industry. Copy number variation (CNV) has been examined in many species and is recognized as a major source of genetic variation that directly contributes to phenotypic variation such as resistance to infectious diseases. Two highly inbred chicken lines, 63 (MD-resistant) and 72 (MD-susceptible), as well as their F1 generation and six recombinant congenic strains (RCSs) with varied susceptibility to MD, are considered as ideal models to identify the complex mechanisms of genetic and molecular resistance to MD.
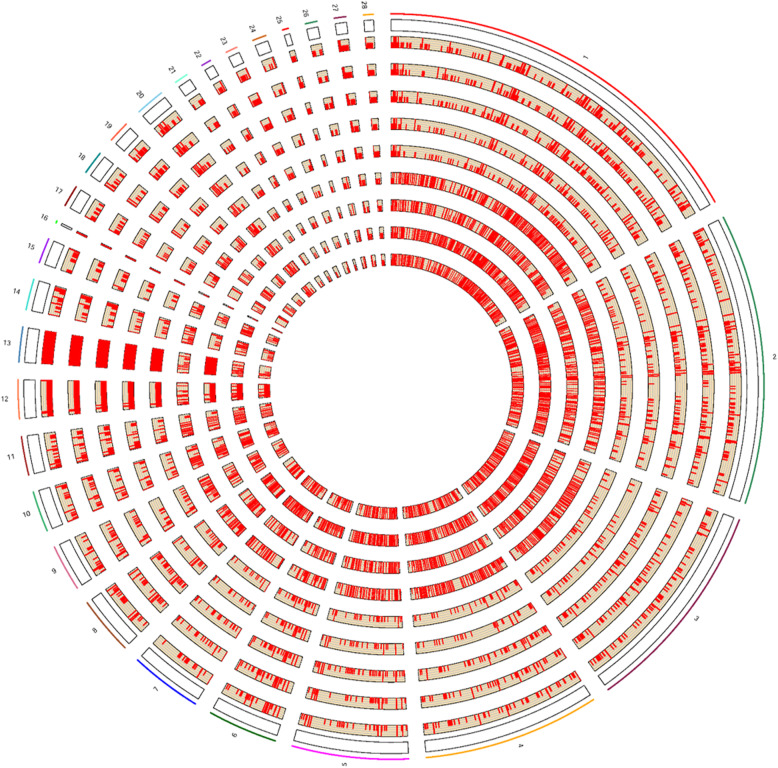
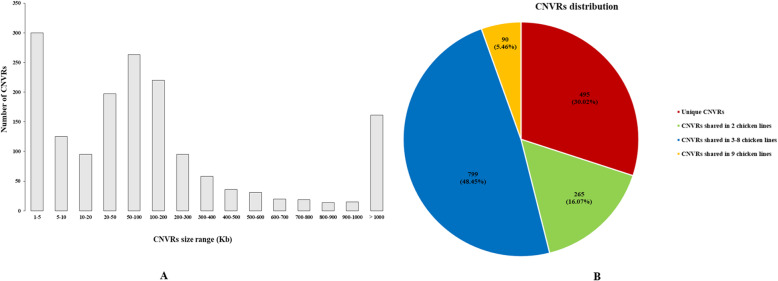
Results: In the present study, to unravel the potential genetic mechanisms underlying resistance to MD, we performed a genome-wide CNV detection using next generation sequencing on the inbred chicken lines with the assistance of CNVnator. As a result, a total of 1649 CNV regions (CNVRs) were successfully identified after merging all the nine datasets, of which 90 CNVRs were overlapped across all the chicken lines. Within these shared regions, 1360 harbored genes were identified. In addition, 55 and 44 CNVRs with 62 and 57 harbored genes were specifically identified in line 63 and 72, respectively. Bioinformatics analysis showed that the nearby genes were significantly enriched in 36 GO terms and 6 KEGG pathways including JAK/STAT signaling pathway. Ten CNVRs (nine deletions and one duplication) involved in 10 disease-related genes were selected for validation by using quantitative real-time PCR (qPCR), all of which were successfully confirmed. Finally, qPCR was also used to validate two deletion events in line 72 that were definitely normal in line 63. One high-confidence gene, IRF2 was identified as the most promising candidate gene underlying resistance and susceptibility to MD in view of its function and overlaps with data from previous study.
Conclusions: Our findings provide valuable insights for understanding the genetic mechanism of resistance to MD and the identified gene and pathway could be considered as the subject of further functional characterization.
期刊介绍:
BMC Genetics is an open access, peer-reviewed journal that considers articles on all aspects of inheritance and variation in individuals and among populations.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
