Jarosław Kończak, Bartosz Janusz, Jakub Młokosiewicz, Tadeusz Satława, Sonia Wróbel, Paweł Dudzic, Konrad Krawczyk
{"title":"Structural pre-training improves physical accuracy of antibody structure prediction using deep learning.","authors":"Jarosław Kończak, Bartosz Janusz, Jakub Młokosiewicz, Tadeusz Satława, Sonia Wróbel, Paweł Dudzic, Konrad Krawczyk","doi":"10.1016/j.immuno.2023.100028","DOIUrl":null,"url":null,"abstract":"<div><p>Protein folding problem obtained a practical solution recently, owing to advances in deep learning. There are classes of proteins though, such as antibodies, that are structurally unique, where the general solution still lacks. In particular, the prediction of the CDR-H3 loop, which is an instrumental part of an antibody in its antigen recognition abilities, remains a challenge. Antibody-specific deep learning frameworks were proposed to tackle this problem noting great progress, both on accuracy and speed fronts. Oftentimes though, the original networks produce physically implausible bond geometries that then need to undergo a time-consuming energy minimization process. Here we hypothesized that pre-training the network on a large, augmented set of models with correct physical geometries, rather than a small set of real antibody X-ray structures, would allow the network to learn better bond geometries. We show that fine-tuning such a pre-trained network on a task of shape prediction on real X-ray structures improves the number of correct peptide bond distances, abstracted as the Cα distances. We further demonstrate that pre-training allows the network to produce physically plausible shapes on an artificial set of CDR-H3s, showing the ability to generalize to the vast antibody sequence space. We hope that our strategy will benefit the development of deep learning antibody models that rapidly generate physically plausible geometries, without the burden of time-consuming energy minimization.</p></div>","PeriodicalId":73343,"journal":{"name":"Immunoinformatics (Amsterdam, Netherlands)","volume":"11 ","pages":"Article 100028"},"PeriodicalIF":0.0000,"publicationDate":"2023-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Immunoinformatics (Amsterdam, Netherlands)","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2667119023000083","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
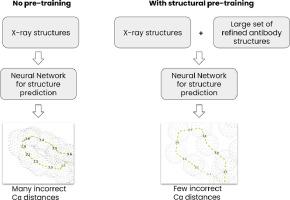
Protein folding problem obtained a practical solution recently, owing to advances in deep learning. There are classes of proteins though, such as antibodies, that are structurally unique, where the general solution still lacks. In particular, the prediction of the CDR-H3 loop, which is an instrumental part of an antibody in its antigen recognition abilities, remains a challenge. Antibody-specific deep learning frameworks were proposed to tackle this problem noting great progress, both on accuracy and speed fronts. Oftentimes though, the original networks produce physically implausible bond geometries that then need to undergo a time-consuming energy minimization process. Here we hypothesized that pre-training the network on a large, augmented set of models with correct physical geometries, rather than a small set of real antibody X-ray structures, would allow the network to learn better bond geometries. We show that fine-tuning such a pre-trained network on a task of shape prediction on real X-ray structures improves the number of correct peptide bond distances, abstracted as the Cα distances. We further demonstrate that pre-training allows the network to produce physically plausible shapes on an artificial set of CDR-H3s, showing the ability to generalize to the vast antibody sequence space. We hope that our strategy will benefit the development of deep learning antibody models that rapidly generate physically plausible geometries, without the burden of time-consuming energy minimization.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
