{"title":"单细胞RNA测序鉴定了与脑膜瘤临床特征和肿瘤微环境相关的巨噬细胞特征。","authors":"Xiaowei Zhang","doi":"10.1049/syb2.12074","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Background</h3>\n \n <p>Meningiomas are common primary brain tumours, with macrophages playing a crucial role in their development and progression. This study aims to identify module genes correlated with meningioma-associated macrophages and analyse their correlation with clinical features and immune infiltration.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>We analysed single-cell RNA sequencing (scRNA-seq) data from two paired meningioma and normal meninges to identify meningioma-associated macrophages. High-dimensional weighted gene co-expression network analysis (hdWGCNA) was employed to identify module genes linked to these macrophages, followed by functional enrichment and pseudotime trajectory analyses. A machine learning-based model using the module genes was developed to predict tumour grades. Finally, meningiomas were classified into two molecular subtypes based on the module genes, followed by a comparison of clinical characteristics and immune cell infiltration.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>Meningiomas exhibited a significantly higher proportion of macrophages than normal meninges, including novel macrophage clusters referred to as meningioma-associated macrophages. The hdWGCNA analysis of macrophages within meningiomas unveiled 12 distinct modules, with the blue, black, and turquoise modules closely correlated with the meningioma-associated macrophages. Hub genes within these modules were enriched in immune regulation, cellular communication, and metabolism pathways. Machine learning analysis identified 13 module genes (RSBN1, TIPRL, ATIC, SPP1, MALSU1, CDK1, MGP, DDIT3, SUPT16H, NFKBIA, SRSF5, ATXN2L, and UBB) strongly correlated with meningioma grade and constructed a predictive model with high accuracy and robustness. Based on the module genes, meningiomas were classified into two subtypes with distinct clinical and tumour microenvironment characteristics.</p>\n </section>\n \n <section>\n \n <h3> Conclusions</h3>\n \n <p>Our findings provide insights into the molecular characteristics underlying macrophage infiltration in meningiomas. The molecular signatures of macrophages demonstrate correlations with clinical features and immune cell infiltration in meningiomas.</p>\n </section>\n </div>","PeriodicalId":50379,"journal":{"name":"IET Systems Biology","volume":"17 5","pages":"259-270"},"PeriodicalIF":1.9000,"publicationDate":"2023-07-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ietresearch.onlinelibrary.wiley.com/doi/epdf/10.1049/syb2.12074","citationCount":"0","resultStr":"{\"title\":\"Single-cell RNA sequencing identifies macrophage signatures correlated with clinical features and tumour microenvironment in meningiomas\",\"authors\":\"Xiaowei Zhang\",\"doi\":\"10.1049/syb2.12074\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n \\n <section>\\n \\n <h3> Background</h3>\\n \\n <p>Meningiomas are common primary brain tumours, with macrophages playing a crucial role in their development and progression. This study aims to identify module genes correlated with meningioma-associated macrophages and analyse their correlation with clinical features and immune infiltration.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Methods</h3>\\n \\n <p>We analysed single-cell RNA sequencing (scRNA-seq) data from two paired meningioma and normal meninges to identify meningioma-associated macrophages. High-dimensional weighted gene co-expression network analysis (hdWGCNA) was employed to identify module genes linked to these macrophages, followed by functional enrichment and pseudotime trajectory analyses. A machine learning-based model using the module genes was developed to predict tumour grades. Finally, meningiomas were classified into two molecular subtypes based on the module genes, followed by a comparison of clinical characteristics and immune cell infiltration.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Results</h3>\\n \\n <p>Meningiomas exhibited a significantly higher proportion of macrophages than normal meninges, including novel macrophage clusters referred to as meningioma-associated macrophages. The hdWGCNA analysis of macrophages within meningiomas unveiled 12 distinct modules, with the blue, black, and turquoise modules closely correlated with the meningioma-associated macrophages. Hub genes within these modules were enriched in immune regulation, cellular communication, and metabolism pathways. Machine learning analysis identified 13 module genes (RSBN1, TIPRL, ATIC, SPP1, MALSU1, CDK1, MGP, DDIT3, SUPT16H, NFKBIA, SRSF5, ATXN2L, and UBB) strongly correlated with meningioma grade and constructed a predictive model with high accuracy and robustness. Based on the module genes, meningiomas were classified into two subtypes with distinct clinical and tumour microenvironment characteristics.</p>\\n </section>\\n \\n <section>\\n \\n <h3> Conclusions</h3>\\n \\n <p>Our findings provide insights into the molecular characteristics underlying macrophage infiltration in meningiomas. The molecular signatures of macrophages demonstrate correlations with clinical features and immune cell infiltration in meningiomas.</p>\\n </section>\\n </div>\",\"PeriodicalId\":50379,\"journal\":{\"name\":\"IET Systems Biology\",\"volume\":\"17 5\",\"pages\":\"259-270\"},\"PeriodicalIF\":1.9000,\"publicationDate\":\"2023-07-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ietresearch.onlinelibrary.wiley.com/doi/epdf/10.1049/syb2.12074\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"IET Systems Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1049/syb2.12074\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"IET Systems Biology","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1049/syb2.12074","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
Single-cell RNA sequencing identifies macrophage signatures correlated with clinical features and tumour microenvironment in meningiomas
Background
Meningiomas are common primary brain tumours, with macrophages playing a crucial role in their development and progression. This study aims to identify module genes correlated with meningioma-associated macrophages and analyse their correlation with clinical features and immune infiltration.
Methods
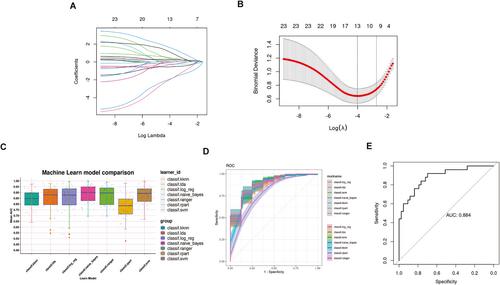
We analysed single-cell RNA sequencing (scRNA-seq) data from two paired meningioma and normal meninges to identify meningioma-associated macrophages. High-dimensional weighted gene co-expression network analysis (hdWGCNA) was employed to identify module genes linked to these macrophages, followed by functional enrichment and pseudotime trajectory analyses. A machine learning-based model using the module genes was developed to predict tumour grades. Finally, meningiomas were classified into two molecular subtypes based on the module genes, followed by a comparison of clinical characteristics and immune cell infiltration.
Results
Meningiomas exhibited a significantly higher proportion of macrophages than normal meninges, including novel macrophage clusters referred to as meningioma-associated macrophages. The hdWGCNA analysis of macrophages within meningiomas unveiled 12 distinct modules, with the blue, black, and turquoise modules closely correlated with the meningioma-associated macrophages. Hub genes within these modules were enriched in immune regulation, cellular communication, and metabolism pathways. Machine learning analysis identified 13 module genes (RSBN1, TIPRL, ATIC, SPP1, MALSU1, CDK1, MGP, DDIT3, SUPT16H, NFKBIA, SRSF5, ATXN2L, and UBB) strongly correlated with meningioma grade and constructed a predictive model with high accuracy and robustness. Based on the module genes, meningiomas were classified into two subtypes with distinct clinical and tumour microenvironment characteristics.
Conclusions
Our findings provide insights into the molecular characteristics underlying macrophage infiltration in meningiomas. The molecular signatures of macrophages demonstrate correlations with clinical features and immune cell infiltration in meningiomas.
期刊介绍:
IET Systems Biology covers intra- and inter-cellular dynamics, using systems- and signal-oriented approaches. Papers that analyse genomic data in order to identify variables and basic relationships between them are considered if the results provide a basis for mathematical modelling and simulation of cellular dynamics. Manuscripts on molecular and cell biological studies are encouraged if the aim is a systems approach to dynamic interactions within and between cells.
The scope includes the following topics:
Genomics, transcriptomics, proteomics, metabolomics, cells, tissue and the physiome; molecular and cellular interaction, gene, cell and protein function; networks and pathways; metabolism and cell signalling; dynamics, regulation and control; systems, signals, and information; experimental data analysis; mathematical modelling, simulation and theoretical analysis; biological modelling, simulation, prediction and control; methodologies, databases, tools and algorithms for modelling and simulation; modelling, analysis and control of biological networks; synthetic biology and bioengineering based on systems biology.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
