Hassan Karami, Samira Nomiri, Mohammad Ghasemigol, Niloufar Mehrvarzian, Afshin Derakhshani, Mohammad Fereidouni, Masoud Mirimoghaddam, Hossein Safarpour
{"title":"CHAC1作为区分脱发与其他皮肤病及判断其严重程度的新生物标志物","authors":"Hassan Karami, Samira Nomiri, Mohammad Ghasemigol, Niloufar Mehrvarzian, Afshin Derakhshani, Mohammad Fereidouni, Masoud Mirimoghaddam, Hossein Safarpour","doi":"10.1049/syb2.12048","DOIUrl":null,"url":null,"abstract":"<p>Alopecia Areata (AA) is characterised by an autoimmune response to hair follicles (HFs) and its exact pathobiology remains unclear. The current study aims to look into the molecular changes in the skin of AA patients as well as the potential underlying molecular mechanisms of AA in order to identify potential candidates for early detection and treatment of AA. We applied Weighted Gene Co-expression Network Analysis (WGCNA) to identify key modules, hub genes, and mRNA–miRNA regulatory networks associated with AA. Furthermore, Chi2 as a machine-learning algorithm was used to compute the gene importance in AA. Finally, drug-target construction revealed the potential of repositioning drugs for the treatment of AA. Our analysis using four AA data sets established a network strongly correlated to AA pathogenicity based on <i>GZMA</i>, <i>OXCT2</i>, <i>HOXC13</i>, <i>KRT40</i>, <i>COMP</i>, <i>CHAC1</i>, and <i>KRT83</i> hub genes. Interestingly, machine learning introduced these genes as important in AA pathogenicity. Besides that, using another ten data sets, we showed that <i>CHAC1</i> could clearly distinguish AA from similar clinical phenotypes, such as scarring alopecia due to psoriasis. Also, two FDA-approved drug candidates and 30 experimentally validated miRNAs were identified that affected the co-expression network. Using transcriptome analysis, suggested <i>CHAC1</i> as a potential diagnostic predictor to diagnose AA.</p>","PeriodicalId":50379,"journal":{"name":"IET Systems Biology","volume":"16 5","pages":"173-185"},"PeriodicalIF":1.9000,"publicationDate":"2022-08-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9469792/pdf/","citationCount":"1","resultStr":"{\"title\":\"CHAC1 as a novel biomarker for distinguishing alopecia from other dermatological diseases and determining its severity\",\"authors\":\"Hassan Karami, Samira Nomiri, Mohammad Ghasemigol, Niloufar Mehrvarzian, Afshin Derakhshani, Mohammad Fereidouni, Masoud Mirimoghaddam, Hossein Safarpour\",\"doi\":\"10.1049/syb2.12048\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Alopecia Areata (AA) is characterised by an autoimmune response to hair follicles (HFs) and its exact pathobiology remains unclear. The current study aims to look into the molecular changes in the skin of AA patients as well as the potential underlying molecular mechanisms of AA in order to identify potential candidates for early detection and treatment of AA. We applied Weighted Gene Co-expression Network Analysis (WGCNA) to identify key modules, hub genes, and mRNA–miRNA regulatory networks associated with AA. Furthermore, Chi2 as a machine-learning algorithm was used to compute the gene importance in AA. Finally, drug-target construction revealed the potential of repositioning drugs for the treatment of AA. Our analysis using four AA data sets established a network strongly correlated to AA pathogenicity based on <i>GZMA</i>, <i>OXCT2</i>, <i>HOXC13</i>, <i>KRT40</i>, <i>COMP</i>, <i>CHAC1</i>, and <i>KRT83</i> hub genes. Interestingly, machine learning introduced these genes as important in AA pathogenicity. Besides that, using another ten data sets, we showed that <i>CHAC1</i> could clearly distinguish AA from similar clinical phenotypes, such as scarring alopecia due to psoriasis. Also, two FDA-approved drug candidates and 30 experimentally validated miRNAs were identified that affected the co-expression network. Using transcriptome analysis, suggested <i>CHAC1</i> as a potential diagnostic predictor to diagnose AA.</p>\",\"PeriodicalId\":50379,\"journal\":{\"name\":\"IET Systems Biology\",\"volume\":\"16 5\",\"pages\":\"173-185\"},\"PeriodicalIF\":1.9000,\"publicationDate\":\"2022-08-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9469792/pdf/\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"IET Systems Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1049/syb2.12048\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"IET Systems Biology","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1049/syb2.12048","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
CHAC1 as a novel biomarker for distinguishing alopecia from other dermatological diseases and determining its severity
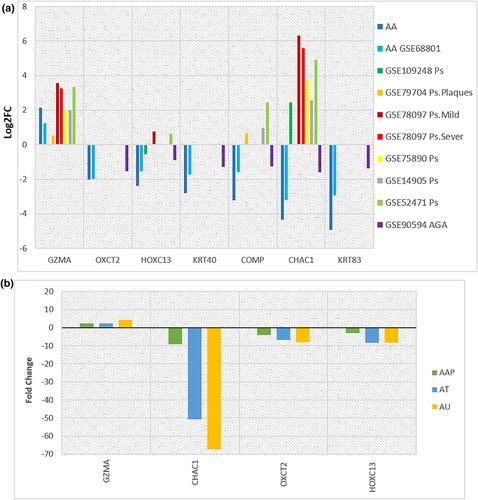
Alopecia Areata (AA) is characterised by an autoimmune response to hair follicles (HFs) and its exact pathobiology remains unclear. The current study aims to look into the molecular changes in the skin of AA patients as well as the potential underlying molecular mechanisms of AA in order to identify potential candidates for early detection and treatment of AA. We applied Weighted Gene Co-expression Network Analysis (WGCNA) to identify key modules, hub genes, and mRNA–miRNA regulatory networks associated with AA. Furthermore, Chi2 as a machine-learning algorithm was used to compute the gene importance in AA. Finally, drug-target construction revealed the potential of repositioning drugs for the treatment of AA. Our analysis using four AA data sets established a network strongly correlated to AA pathogenicity based on GZMA, OXCT2, HOXC13, KRT40, COMP, CHAC1, and KRT83 hub genes. Interestingly, machine learning introduced these genes as important in AA pathogenicity. Besides that, using another ten data sets, we showed that CHAC1 could clearly distinguish AA from similar clinical phenotypes, such as scarring alopecia due to psoriasis. Also, two FDA-approved drug candidates and 30 experimentally validated miRNAs were identified that affected the co-expression network. Using transcriptome analysis, suggested CHAC1 as a potential diagnostic predictor to diagnose AA.
期刊介绍:
IET Systems Biology covers intra- and inter-cellular dynamics, using systems- and signal-oriented approaches. Papers that analyse genomic data in order to identify variables and basic relationships between them are considered if the results provide a basis for mathematical modelling and simulation of cellular dynamics. Manuscripts on molecular and cell biological studies are encouraged if the aim is a systems approach to dynamic interactions within and between cells.
The scope includes the following topics:
Genomics, transcriptomics, proteomics, metabolomics, cells, tissue and the physiome; molecular and cellular interaction, gene, cell and protein function; networks and pathways; metabolism and cell signalling; dynamics, regulation and control; systems, signals, and information; experimental data analysis; mathematical modelling, simulation and theoretical analysis; biological modelling, simulation, prediction and control; methodologies, databases, tools and algorithms for modelling and simulation; modelling, analysis and control of biological networks; synthetic biology and bioengineering based on systems biology.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
