Matteo Castagnola, Rosario Roberto Riso, Alberto Barlini, Enrico Ronca, Henrik Koch
{"title":"精确和近似波函数的极性响应理论","authors":"Matteo Castagnola, Rosario Roberto Riso, Alberto Barlini, Enrico Ronca, Henrik Koch","doi":"10.1002/wcms.1684","DOIUrl":null,"url":null,"abstract":"<p><i>Polaritonic chemistry</i> is an interdisciplinary emerging field that presents several challenges and opportunities in chemistry, physics, and engineering. A systematic review of polaritonic response theory is presented, following a chemical perspective based on molecular response theory. We provide the reader with a general strategy for developing response theory for <i>ab initio</i> cavity quantum electrodynamics (QED) methods and critically emphasize details that still need clarification and require cooperation between the physical and chemistry communities. We show that several well-established results can be applied to strong coupling light-matter systems, leading to novel perspectives on the computation of matter and photonic properties. The application of the Pauli–Fierz Hamiltonian to polaritons is discussed, focusing on the effects of describing operators in different mathematical representations. We thoroughly examine the most common approximations employed in <i>ab initio</i> QED, such as the dipole approximation. We introduce the polaritonic response equations for the recently developed <i>ab initio</i> QED Hartree–Fock and QED coupled cluster methods. The discussion focuses on the similarities and differences from standard quantum chemistry methods, providing practical equations for computing the polaritonic properties.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"14 1","pages":""},"PeriodicalIF":27.0000,"publicationDate":"2023-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1684","citationCount":"0","resultStr":"{\"title\":\"Polaritonic response theory for exact and approximate wave functions\",\"authors\":\"Matteo Castagnola, Rosario Roberto Riso, Alberto Barlini, Enrico Ronca, Henrik Koch\",\"doi\":\"10.1002/wcms.1684\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><i>Polaritonic chemistry</i> is an interdisciplinary emerging field that presents several challenges and opportunities in chemistry, physics, and engineering. A systematic review of polaritonic response theory is presented, following a chemical perspective based on molecular response theory. We provide the reader with a general strategy for developing response theory for <i>ab initio</i> cavity quantum electrodynamics (QED) methods and critically emphasize details that still need clarification and require cooperation between the physical and chemistry communities. We show that several well-established results can be applied to strong coupling light-matter systems, leading to novel perspectives on the computation of matter and photonic properties. The application of the Pauli–Fierz Hamiltonian to polaritons is discussed, focusing on the effects of describing operators in different mathematical representations. We thoroughly examine the most common approximations employed in <i>ab initio</i> QED, such as the dipole approximation. We introduce the polaritonic response equations for the recently developed <i>ab initio</i> QED Hartree–Fock and QED coupled cluster methods. The discussion focuses on the similarities and differences from standard quantum chemistry methods, providing practical equations for computing the polaritonic properties.</p><p>This article is categorized under:\\n </p>\",\"PeriodicalId\":236,\"journal\":{\"name\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"volume\":\"14 1\",\"pages\":\"\"},\"PeriodicalIF\":27.0000,\"publicationDate\":\"2023-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1684\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Wiley Interdisciplinary Reviews: Computational Molecular Science\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://wires.onlinelibrary.wiley.com/doi/10.1002/wcms.1684\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://wires.onlinelibrary.wiley.com/doi/10.1002/wcms.1684","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
摘要
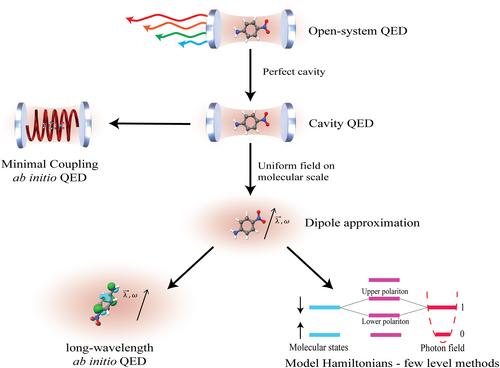
极性化学是一个跨学科的新兴领域,为化学、物理学和工程学带来了诸多挑战和机遇。本文从基于分子响应理论的化学视角出发,对极性响应理论进行了系统综述。我们为读者提供了为自证空穴量子电动力学(QED)方法开发响应理论的一般策略,并批判性地强调了仍需澄清并需要物理和化学界合作的细节。我们表明,一些成熟的结果可以应用于强耦合光-物质系统,从而为物质和光子特性的计算带来新的视角。我们讨论了将保利-费尔茨哈密顿应用于极化子的问题,重点是以不同数学表示法描述算子的效果。我们深入研究了 ab initio QED 中最常用的近似方法,如偶极子近似。我们介绍了最近开发的 ab initio QED 哈特里-福克和 QED 耦合簇方法的极化子响应方程。讨论的重点是与标准量子化学方法的异同,并提供计算极性的实用方程:
Polaritonic response theory for exact and approximate wave functions
Polaritonic chemistry is an interdisciplinary emerging field that presents several challenges and opportunities in chemistry, physics, and engineering. A systematic review of polaritonic response theory is presented, following a chemical perspective based on molecular response theory. We provide the reader with a general strategy for developing response theory for ab initio cavity quantum electrodynamics (QED) methods and critically emphasize details that still need clarification and require cooperation between the physical and chemistry communities. We show that several well-established results can be applied to strong coupling light-matter systems, leading to novel perspectives on the computation of matter and photonic properties. The application of the Pauli–Fierz Hamiltonian to polaritons is discussed, focusing on the effects of describing operators in different mathematical representations. We thoroughly examine the most common approximations employed in ab initio QED, such as the dipole approximation. We introduce the polaritonic response equations for the recently developed ab initio QED Hartree–Fock and QED coupled cluster methods. The discussion focuses on the similarities and differences from standard quantum chemistry methods, providing practical equations for computing the polaritonic properties.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
